

Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication
- PMID: 26819306
- PMCID: PMC4810536
- DOI: 10.1128/JVI.02714-15
Human Cytomegalovirus pTRS1 and pIRS1 Antagonize Protein Kinase R To Facilitate Virus Replication
Abstract
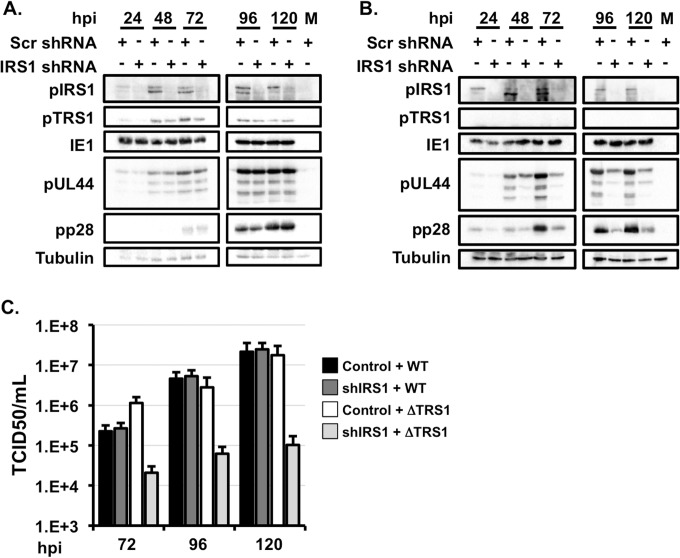
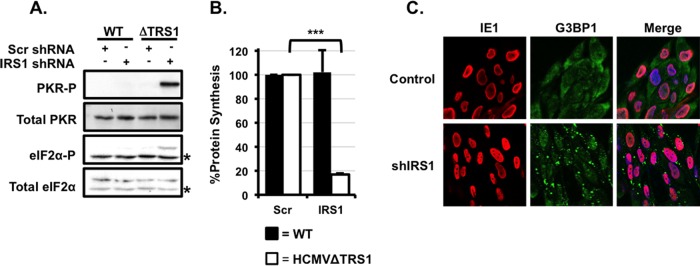
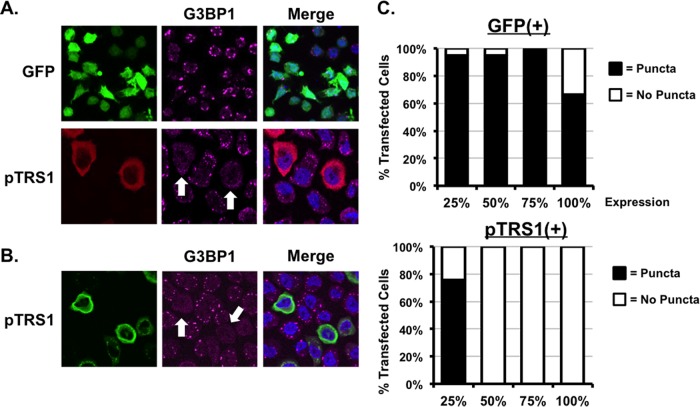
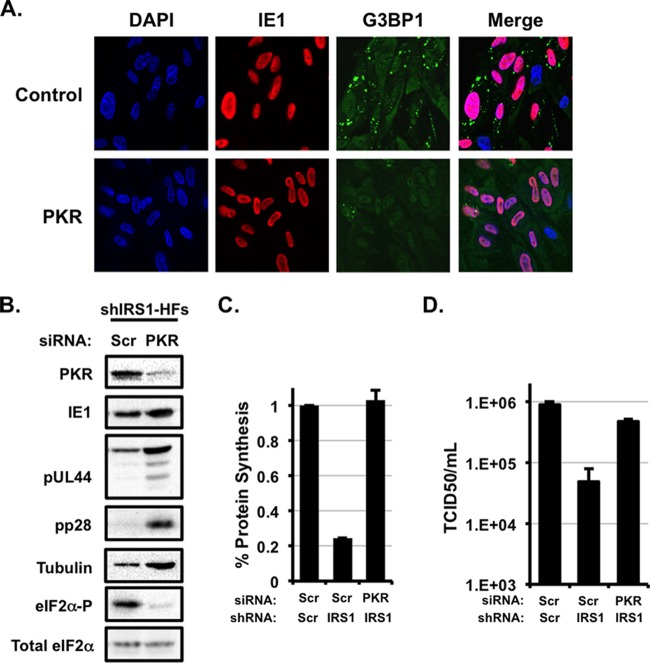
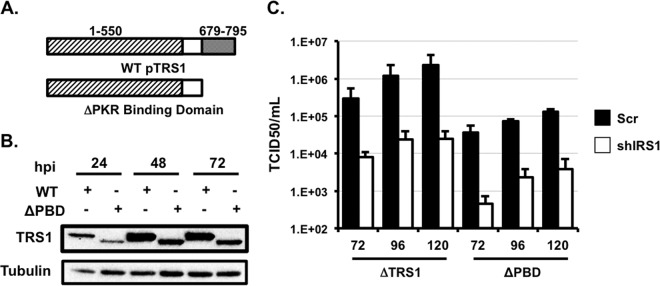
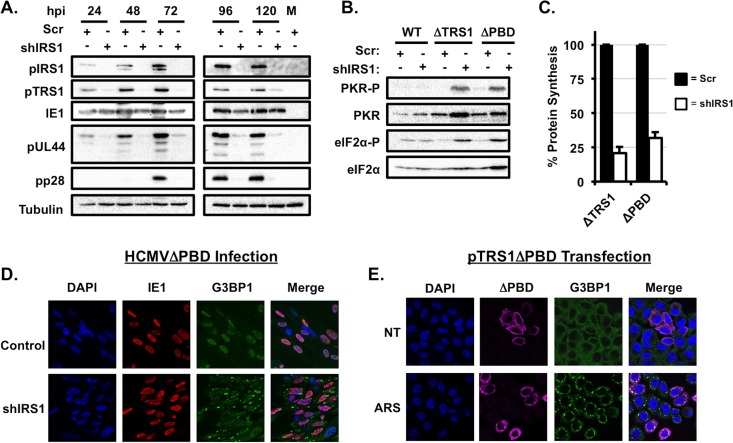
Human cytomegalovirus (HCMV) counteracts host defenses that otherwise act to limit viral protein synthesis. One such defense is the antiviral kinase protein kinase R (PKR), which inactivates the eukaryotic initiation factor 2 (eIF2) translation initiation factor upon binding to viral double-stranded RNAs. Previously, the viral TRS1 and IRS1 proteins were found to antagonize the antiviral kinase PKR outside the context of HCMV infection, and the expression of either pTRS1 or pIRS1 was shown to be necessary for HCMV replication. In this study, we found that expression of either pTRS1 or pIRS1 is necessary to prevent PKR activation during HCMV infection and that antagonism of PKR is critical for efficient viral replication. Consistent with a previous study, we observed decreased overall levels of protein synthesis, reduced viral protein expression, and diminished virus replication in the absence of both pTRS1 and pIRS1. In addition, both PKR and eIF2α were phosphorylated during infection when pTRS1 and pIRS1 were absent. We also found that expression of pTRS1 was both necessary and sufficient to prevent stress granule formation in response to eIF2α phosphorylation. Depletion of PKR prevented eIF2α phosphorylation, rescued HCMV replication and protein synthesis, and reversed the accumulation of stress granules in infected cells. Infection with an HCMV mutant lacking the pTRS1 PKR binding domain resulted in PKR activation, suggesting that pTRS1 inhibits PKR through a direct interaction. Together our results show that antagonism of PKR by HCMV pTRS1 and pIRS1 is critical for viral protein expression and efficient HCMV replication.
Importance: To successfully replicate, viruses must counteract host defenses that limit viral protein synthesis. We have identified inhibition of the antiviral kinase PKR by the viral proteins TRS1 and IRS1 and shown that this is a critical step in HCMV replication. Our results suggest that inhibiting pTRS1 and pIRS1 function or restoring PKR activity during infection may be a successful strategy to limit HCMV disease.
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Protein phosphatase 1 suppresses PKR/EIF2α signaling during human cytomegalovirus infection.J Virol. 2024 Nov 19;98(11):e0059024. doi: 10.1128/jvi.00590-24. Epub 2024 Oct 29. J Virol. 2024. PMID: 39470211
-
Mechanism of Protein Kinase R Inhibition by Human Cytomegalovirus pTRS1.J Virol. 2017 Feb 14;91(5):e01574-16. doi: 10.1128/JVI.01574-16. Print 2017 Mar 1. J Virol. 2017. PMID: 27974558 Free PMC article.
-
Essential role for either TRS1 or IRS1 in human cytomegalovirus replication.J Virol. 2009 May;83(9):4112-20. doi: 10.1128/JVI.02489-08. Epub 2009 Feb 11. J Virol. 2009. PMID: 19211736 Free PMC article.
-
Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase.Pharmacol Ther. 1998 Apr;78(1):29-46. doi: 10.1016/s0163-7258(97)00165-4. Pharmacol Ther. 1998. PMID: 9593328 Review.
-
Viral proteins targeting host protein kinase R to evade an innate immune response: a mini review.Biotechnol Genet Eng Rev. 2018 Apr;34(1):33-59. doi: 10.1080/02648725.2018.1467151. Epub 2018 May 2. Biotechnol Genet Eng Rev. 2018. PMID: 29716441 Review.
Cited by
-
RNA recognition by PKR during DNA virus infection.J Med Virol. 2024 Feb;96(2):e29424. doi: 10.1002/jmv.29424. J Med Virol. 2024. PMID: 38285432 Review.
-
Human cytomegalovirus modulates mTORC1 to redirect mRNA translation within quiescently infected monocytes.J Virol. 2024 Feb 20;98(2):e0188823. doi: 10.1128/jvi.01888-23. Epub 2024 Jan 30. J Virol. 2024. PMID: 38289104 Free PMC article.
-
Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III.Viruses. 2022 Apr 9;14(4):779. doi: 10.3390/v14040779. Viruses. 2022. PMID: 35458509 Free PMC article.
-
Insights into the Transcriptome of Human Cytomegalovirus: A Comprehensive Review.Viruses. 2023 Aug 8;15(8):1703. doi: 10.3390/v15081703. Viruses. 2023. PMID: 37632045 Free PMC article. Review.
-
NLRX1 promotes immediate IRF1-directed antiviral responses by limiting dsRNA-activated translational inhibition mediated by PKR.Nat Immunol. 2017 Dec;18(12):1299-1309. doi: 10.1038/ni.3853. Epub 2017 Oct 2. Nat Immunol. 2017. PMID: 28967880 Free PMC article.
References
-
- Romano PR, Garcia-Barrio MT, Zhang X, Wang Q, Taylor DR, Zhang F, Herring C, Mathews MB, Qin J, Hinnebusch AG. 1998. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2alpha kinases PKR and GCN2. Mol Cell Biol 18:2282–2297. doi:10.1128/MCB.18.4.2282. - DOI - PMC - PubMed
-
- Samuel CE. 1979. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc Natl Acad Sci U S A 76:600–604. doi:10.1073/pnas.76.2.600. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
