

Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy
- PMID: 26718575
- PMCID: PMC4776089
- DOI: 10.1212/WNL.0000000000002324
Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy
Erratum in
-
Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy.Neurology. 2016 Mar 15;86(11):1077. doi: 10.1212/WNL.0000000000002556. Neurology. 2016. PMID: 26976520 Free PMC article. No abstract available.
Abstract
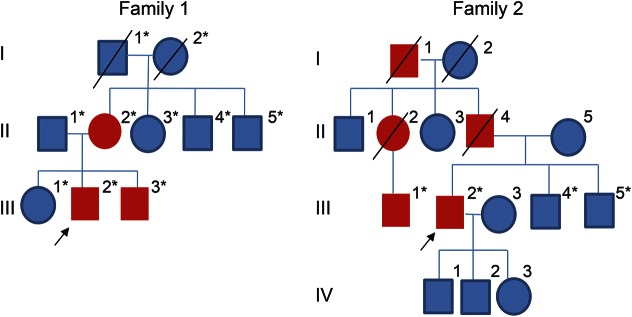
Objective: To report novel disease and pathology due to HSPB8 mutations in 2 families with autosomal dominant distal neuromuscular disease showing both myofibrillar and rimmed vacuolar myopathy together with neurogenic changes.
Methods: We performed whole-exome sequencing (WES) in tandem with linkage analysis and candidate gene approach as well as targeted next-generation sequencing (tNGS) to identify causative mutations in 2 families with dominant rimmed vacuolar myopathy and a motor neuropathy. Pathogenic variants and familial segregation were confirmed using Sanger sequencing.
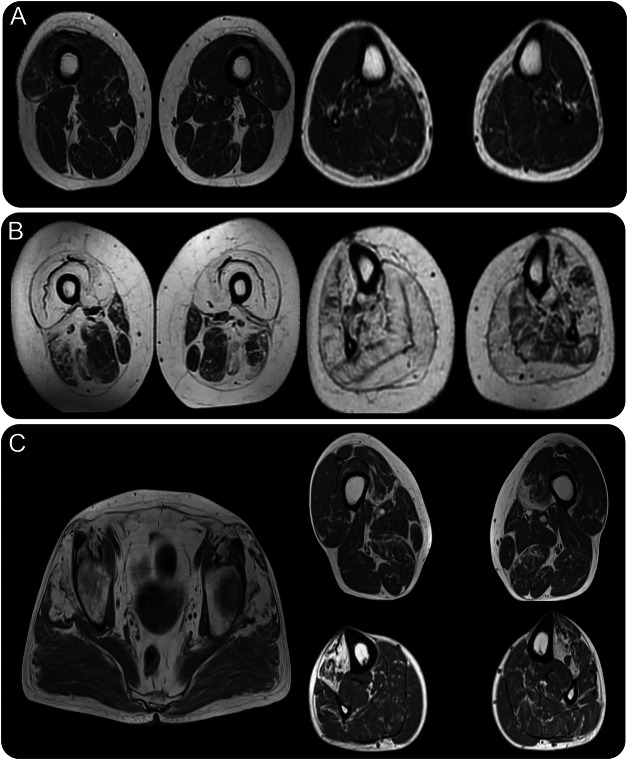
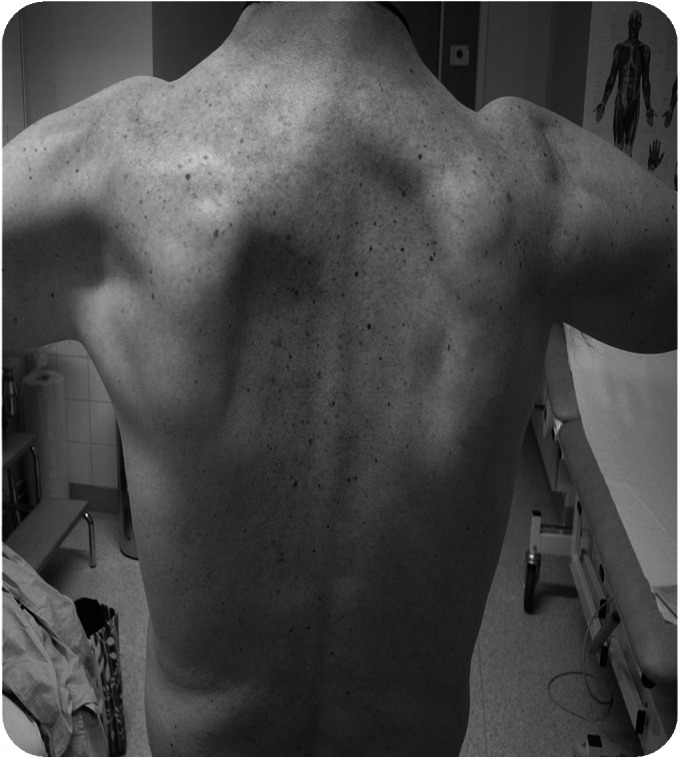
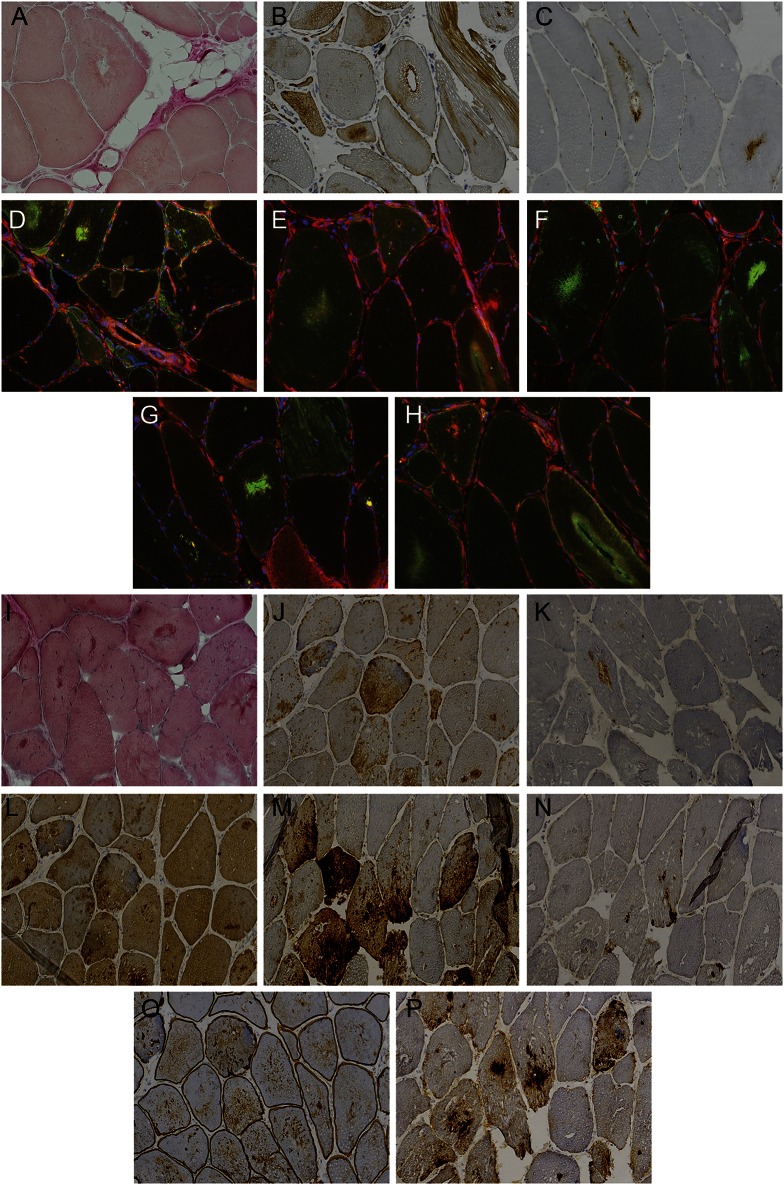
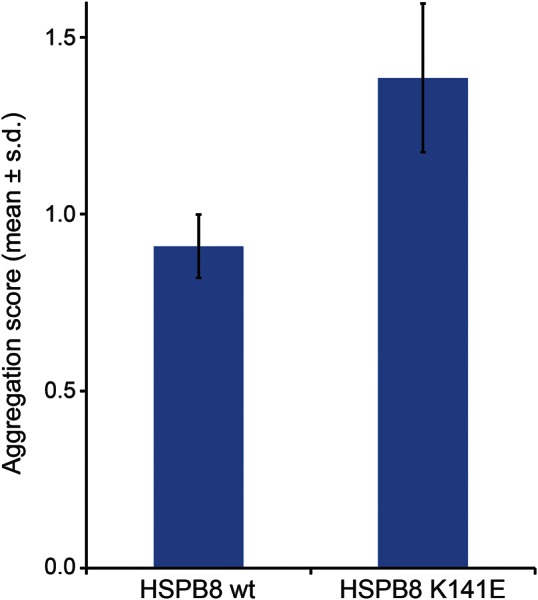
Results: WES and tNGS identified a heterozygous change in HSPB8 in both families: c.421A > G p.K141E in family 1 and c.151insC p.P173SfsX43 in family 2. Affected patients had a distal myopathy that showed myofibrillar aggregates and rimmed vacuoles combined with a clear neurogenic component both on biopsy and neurophysiologic studies. MRI of lower limb muscles demonstrated diffuse tissue changes early in the disease stage progressing later to fatty replacement typical of a myopathy.
Conclusion: We expand the understanding of disease mechanisms, tissue involvement, and phenotypic outcome of HSPB8 mutations. HSPB8 is part of the chaperone-assisted selective autophagy (CASA) complex previously only associated with Charcot-Marie-Tooth type 2L (OMIM 60673) and distal hereditary motor neuronopathy type IIa. However, we now demonstrate that patients can develop a myopathy with histologic features of myofibrillar myopathy with aggregates and rimmed vacuoles, similar to the pathology in myopathies due to gene defects in other compounds of the CASA complex such as BAG3 and DNAJB6 after developing the early neurogenic effects.
© 2015 American Academy of Neurology.
Figures
Similar articles
-
Altered TDP-43-dependent splicing in HSPB8-related distal hereditary motor neuropathy and myofibrillar myopathy.Eur J Neurol. 2018 Jan;25(1):154-163. doi: 10.1111/ene.13478. Epub 2017 Dec 2. Eur J Neurol. 2018. PMID: 29029362
-
A novel heterozygous mutation in the C-terminal region of HSPB8 leads to limb-girdle rimmed vacuolar myopathy.Neuromuscul Disord. 2020 Mar;30(3):236-240. doi: 10.1016/j.nmd.2020.02.005. Epub 2020 Feb 12. Neuromuscul Disord. 2020. PMID: 32165108
-
A novel deletion in the C-terminal region of HSPB8 in a family with rimmed vacuolar myopathy.J Hum Genet. 2021 Oct;66(10):965-972. doi: 10.1038/s10038-021-00916-y. Epub 2021 Mar 20. J Hum Genet. 2021. PMID: 33744911
-
Panorama of the distal myopathies.Acta Myol. 2020 Dec 1;39(4):245-265. doi: 10.36185/2532-1900-028. eCollection 2020 Dec. Acta Myol. 2020. PMID: 33458580 Free PMC article. Review.
-
The distal hereditary motor neuropathies.J Neurol Neurosurg Psychiatry. 2012 Jan;83(1):6-14. doi: 10.1136/jnnp-2011-300952. Epub 2011 Oct 25. J Neurol Neurosurg Psychiatry. 2012. PMID: 22028385 Review.
Cited by
-
RNA-Seq identifies genes whose proteins are upregulated during syncytia development in murine C2C12 myoblasts and human BeWo trophoblasts.Physiol Rep. 2021 Jan;9(1):e14671. doi: 10.14814/phy2.14671. Physiol Rep. 2021. PMID: 33403800 Free PMC article.
-
Differential diagnosis of vacuolar myopathies in the NGS era.Brain Pathol. 2020 Sep;30(5):877-896. doi: 10.1111/bpa.12864. Epub 2020 Jun 15. Brain Pathol. 2020. PMID: 32419263 Free PMC article.
-
Combining enhanced sampling and deep learning dimensionality reduction for the study of the heat shock protein B8 and its pathological mutant K141E.RSC Adv. 2022 Nov 8;12(49):31996-32011. doi: 10.1039/d2ra04913a. eCollection 2022 Nov 3. RSC Adv. 2022. PMID: 36380940 Free PMC article.
-
An interaction study in mammalian cells demonstrates weak binding of HSPB2 to BAG3, which is regulated by HSPB3 and abrogated by HSPB8.Cell Stress Chaperones. 2017 Jul;22(4):531-540. doi: 10.1007/s12192-017-0769-x. Epub 2017 Feb 8. Cell Stress Chaperones. 2017. PMID: 28181153 Free PMC article.
-
The Role of the Heat Shock Protein B8 (HSPB8) in Motoneuron Diseases.Front Mol Neurosci. 2017 Jun 21;10:176. doi: 10.3389/fnmol.2017.00176. eCollection 2017. Front Mol Neurosci. 2017. PMID: 28680390 Free PMC article. Review.
References
-
- Tang B, Zhao GH, Luo W, et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type 2L. Hum Genet 2005;116:222–224. - PubMed
-
- Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat shock protein 22 causes distal motor neuropathy. Nat Genet 2004;36:597–601. - PubMed
-
- Ulbricht A, Eppler FJ, Tapia VE, et al. Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol 2013;23:5. - PubMed
-
- Carra S, Sivilotti M, Chavez Zobel AT, Lambert H, Landry J. HSBP8, a small heat shock protein mutated in human neuromuscular disorders, has in vivo chaperone activity in cultured cells. Hum Mol Genet 2005;14:1659–1669. - PubMed
-
- Arndt V, Dick N, Tawo R, et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr Biol 2010;20:143–148. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous