

Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or 'classical' congenital myopathy
- PMID: 26700687
- PMCID: PMC4766374
- DOI: 10.1093/brain/awv352
Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or 'classical' congenital myopathy
Abstract
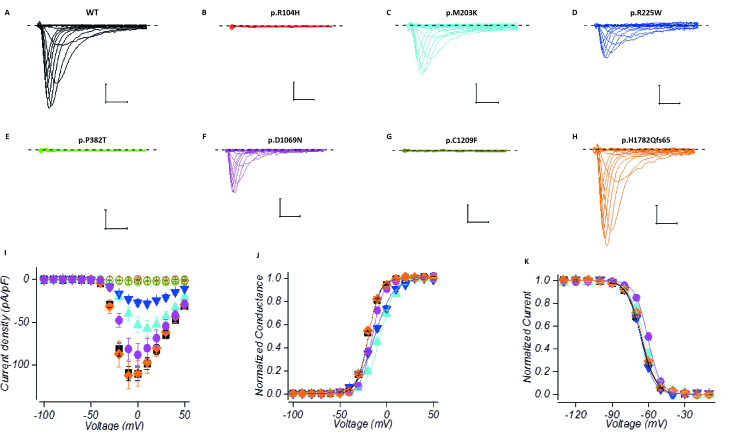
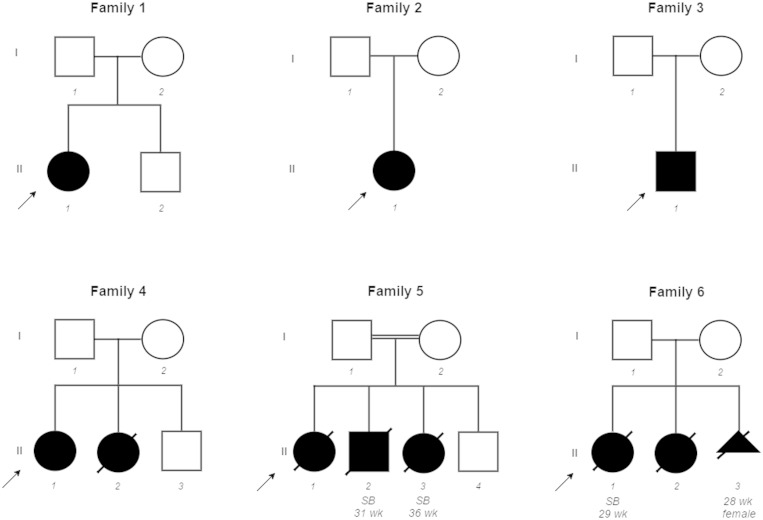
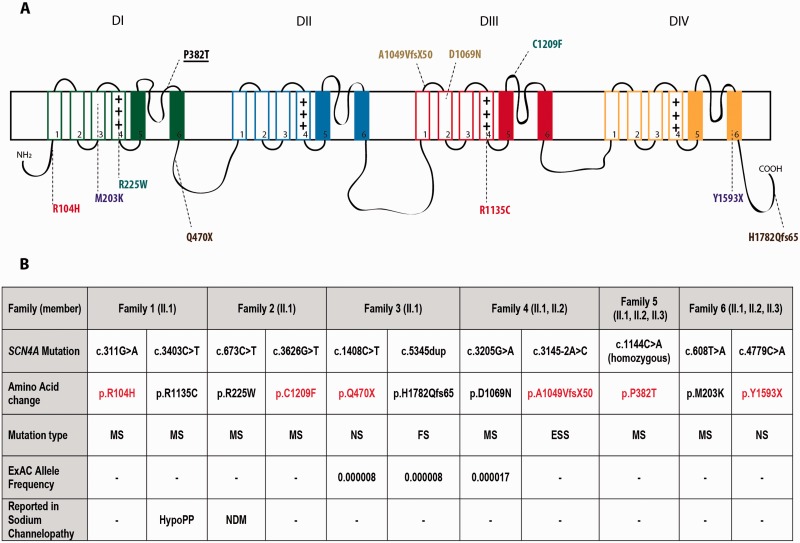
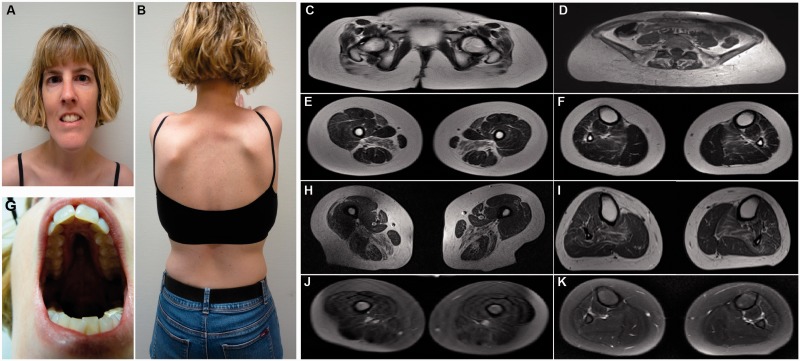
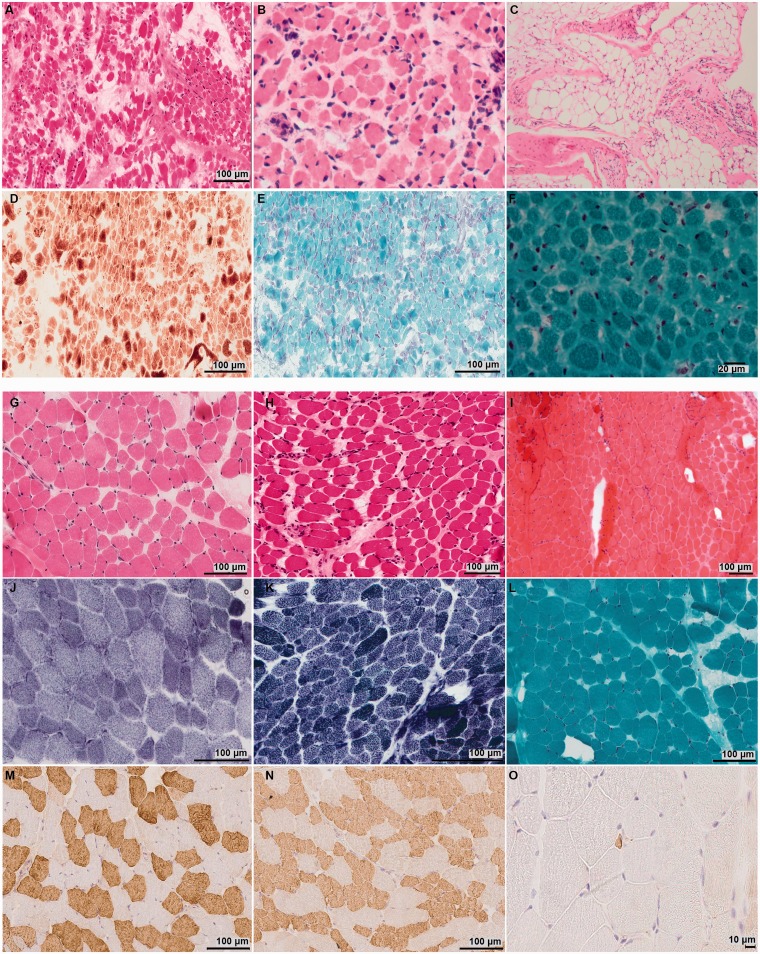
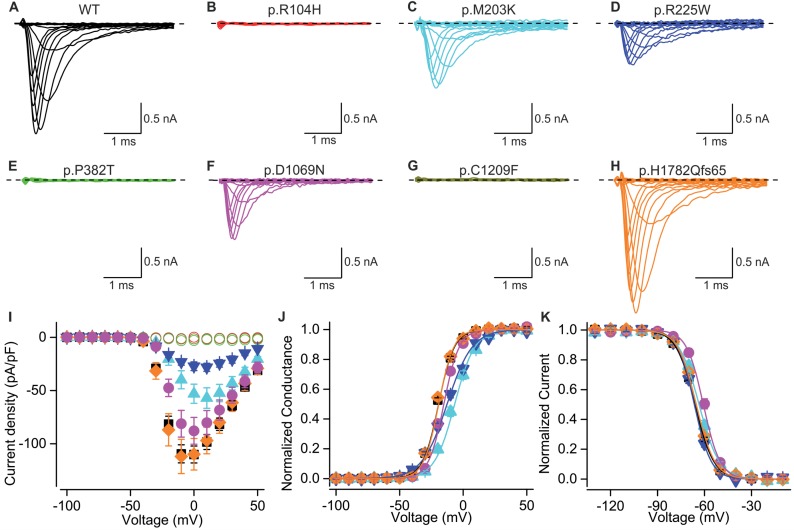
Congenital myopathies are a clinically and genetically heterogeneous group of muscle disorders characterized by congenital or early-onset hypotonia and muscle weakness, and specific pathological features on muscle biopsy. The phenotype ranges from foetal akinesia resulting in in utero or neonatal mortality, to milder disorders that are not life-limiting. Over the past decade, more than 20 new congenital myopathy genes have been identified. Most encode proteins involved in muscle contraction; however, mutations in ion channel-encoding genes are increasingly being recognized as a cause of this group of disorders. SCN4A encodes the α-subunit of the skeletal muscle voltage-gated sodium channel (Nav1.4). This channel is essential for the generation and propagation of the muscle action potential crucial to muscle contraction. Dominant SCN4A gain-of-function mutations are a well-established cause of myotonia and periodic paralysis. Using whole exome sequencing, we identified homozygous or compound heterozygous SCN4A mutations in a cohort of 11 individuals from six unrelated kindreds with congenital myopathy. Affected members developed in utero- or neonatal-onset muscle weakness of variable severity. In seven cases, severe muscle weakness resulted in death during the third trimester or shortly after birth. The remaining four cases had marked congenital or neonatal-onset hypotonia and weakness associated with mild-to-moderate facial and neck weakness, significant neonatal-onset respiratory and swallowing difficulties and childhood-onset spinal deformities. All four surviving cohort members experienced clinical improvement in the first decade of life. Muscle biopsies showed myopathic features including fibre size variability, presence of fibrofatty tissue of varying severity, without specific structural abnormalities. Electrophysiology suggested a myopathic process, without myotonia. In vitro functional assessment in HEK293 cells of the impact of the identified SCN4A mutations showed loss-of-function of the mutant Nav1.4 channels. All, apart from one, of the mutations either caused fully non-functional channels, or resulted in a reduced channel activity. Each of the affected cases carried at least one full loss-of-function mutation. In five out of six families, a second loss-of-function mutation was present on the trans allele. These functional results provide convincing evidence for the pathogenicity of the identified mutations and suggest that different degrees of loss-of-function in mutant Nav1.4 channels are associated with attenuation of the skeletal muscle action potential amplitude to a level insufficient to support normal muscle function. The results demonstrate that recessive loss-of-function SCN4A mutations should be considered in patients with a congenital myopathy.
Keywords: SCN4A; congenital myopathy; foetal akinesia; foetal hypokinesia; loss-of-function mutation.
© The Author (2015). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
When all is lost…a severe myopathy with hypotonia from sodium channel mutations.Brain. 2016 Mar;139(Pt 3):642-4. doi: 10.1093/brain/awv400. Brain. 2016. PMID: 26917582 Free PMC article.
Similar articles
-
When all is lost…a severe myopathy with hypotonia from sodium channel mutations.Brain. 2016 Mar;139(Pt 3):642-4. doi: 10.1093/brain/awv400. Brain. 2016. PMID: 26917582 Free PMC article.
-
A Novel De Novo Heterozygous SCN4a Mutation Causing Congenital Myopathy, Myotonia and Multiple Congenital Anomalies.J Neuromuscul Dis. 2019;6(4):467-473. doi: 10.3233/JND-190425. J Neuromuscul Dis. 2019. PMID: 31609695
-
Myasthenic congenital myopathy from recessive mutations at a single residue in NaV1.4.Neurology. 2019 Mar 26;92(13):e1405-e1415. doi: 10.1212/WNL.0000000000007185. Epub 2019 Mar 1. Neurology. 2019. PMID: 30824560 Free PMC article.
-
Sodium Channelopathies of Skeletal Muscle.Handb Exp Pharmacol. 2018;246:309-330. doi: 10.1007/164_2017_52. Handb Exp Pharmacol. 2018. PMID: 28939973 Free PMC article. Review.
-
New Challenges Resulting From the Loss of Function of Nav1.4 in Neuromuscular Diseases.Front Pharmacol. 2021 Oct 4;12:751095. doi: 10.3389/fphar.2021.751095. eCollection 2021. Front Pharmacol. 2021. PMID: 34671263 Free PMC article. Review.
Cited by
-
Spider toxin inhibits gating pore currents underlying periodic paralysis.Proc Natl Acad Sci U S A. 2018 Apr 24;115(17):4495-4500. doi: 10.1073/pnas.1720185115. Epub 2018 Apr 10. Proc Natl Acad Sci U S A. 2018. PMID: 29636418 Free PMC article.
-
Dysfunction of NaV1.4, a skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study.Lancet. 2018 Apr 14;391(10129):1483-1492. doi: 10.1016/S0140-6736(18)30021-7. Epub 2018 Apr 5. Lancet. 2018. PMID: 29605429 Free PMC article.
-
Expanding the spectrum of congenital myopathy linked to recessive mutations in SCN4A.Neurology. 2017 Jan 24;88(4):414-416. doi: 10.1212/WNL.0000000000003535. Epub 2016 Dec 21. Neurology. 2017. PMID: 28003497 Free PMC article. No abstract available.
-
Sodium Channel Gene Variants in Fetuses with Abnormal Sonographic Findings: Expanding the Prenatal Phenotypic Spectrum of Sodium Channelopathies.Genes (Basel). 2024 Jan 18;15(1):119. doi: 10.3390/genes15010119. Genes (Basel). 2024. PMID: 38255008 Free PMC article.
-
The Increasing Impact of Translational Research in the Molecular Diagnostics of Neuromuscular Diseases.Int J Mol Sci. 2021 Apr 20;22(8):4274. doi: 10.3390/ijms22084274. Int J Mol Sci. 2021. PMID: 33924139 Free PMC article. Review.
References
-
- de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. . Diagnostic exome sequencing in persons with severe intellectual disability . N Engl J Med 2012. ; 367 : 1921 – 9 . - PubMed
-
- Ehmsen J, Poon E, Davies K . The dystrophin-associated protein complex [Review] . J Cell Sci 2002. ; 115 : 2801 – 3 . - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
