

TNF biology, pathogenic mechanisms and emerging therapeutic strategies
- PMID: 26656660
- PMCID: PMC4809675
- DOI: 10.1038/nrrheum.2015.169
TNF biology, pathogenic mechanisms and emerging therapeutic strategies
Abstract
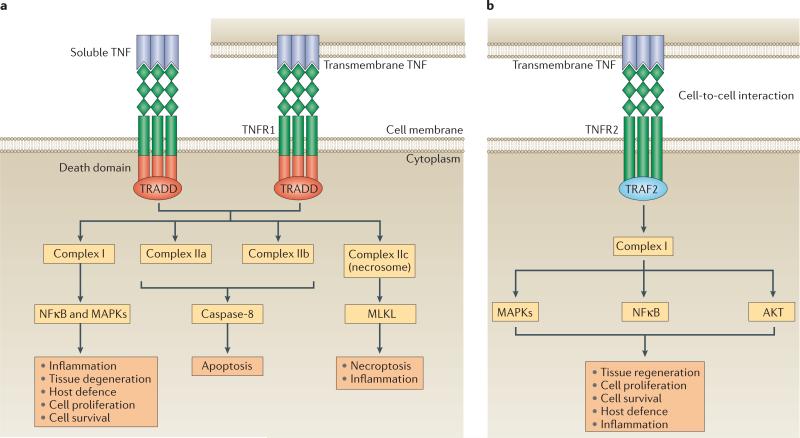
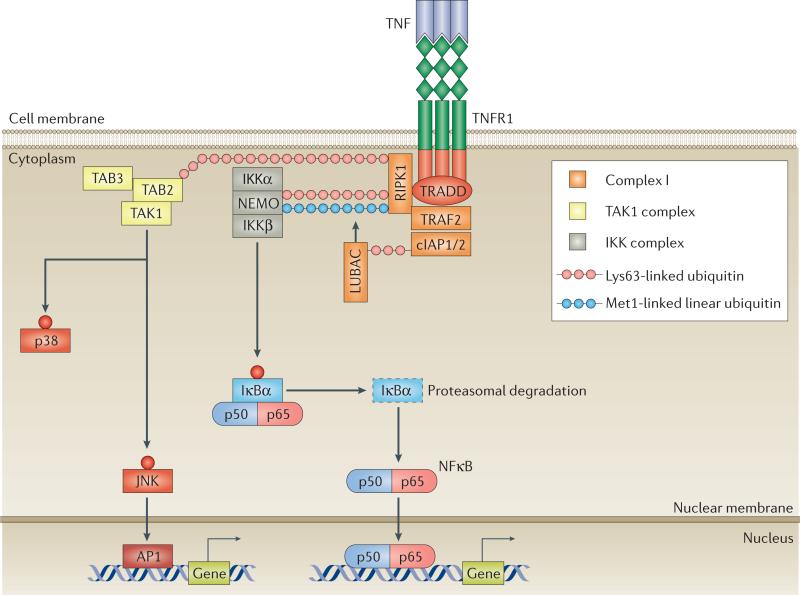
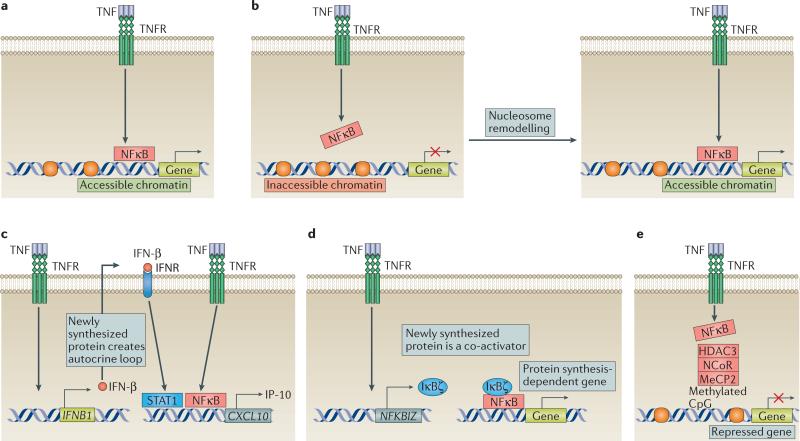
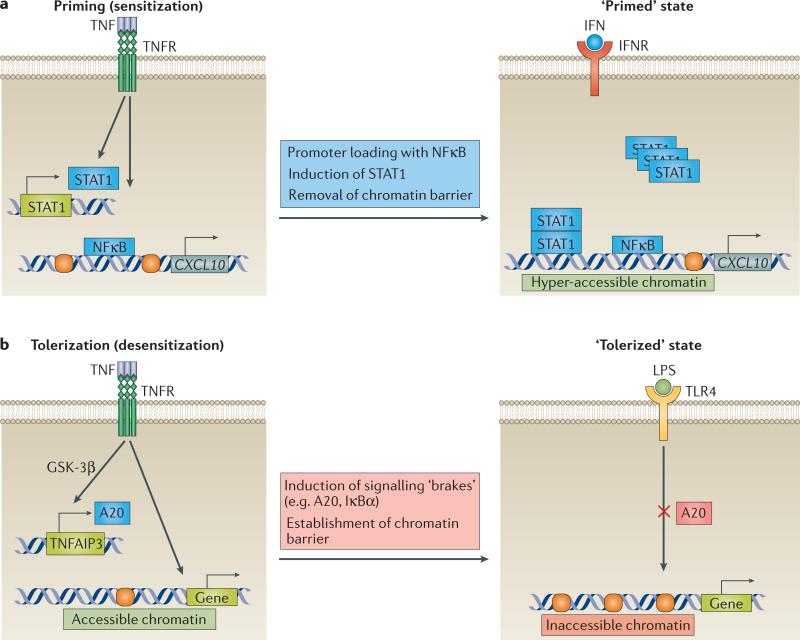
TNF is a pleiotropic cytokine with important functions in homeostasis and disease pathogenesis. Recent discoveries have provided insights into TNF biology that introduce new concepts for the development of therapeutics for TNF-mediated diseases. The model of TNF receptor signalling has been extended to include linear ubiquitination and the formation of distinct signalling complexes that are linked with different functional outcomes, such as inflammation, apoptosis and necroptosis. Our understanding of TNF-induced gene expression has been enriched by the discovery of epigenetic mechanisms and concepts related to cellular priming, tolerization and induction of 'short-term transcriptional memory'. Identification of distinct homeostatic or pathogenic TNF-induced signalling pathways has introduced the concept of selectively inhibiting the deleterious effects of TNF while preserving its homeostatic bioactivities for therapeutic purposes. In this Review, we present molecular mechanisms underlying the roles of TNF in homeostasis and inflammatory disease pathogenesis, and discuss novel strategies to advance therapeutic paradigms for the treatment of TNF-mediated diseases.
Figures
Similar articles
-
TNF-α promotes nuclear enrichment of the transcription factor TonEBP/NFAT5 to selectively control inflammatory but not osmoregulatory responses in nucleus pulposus cells.J Biol Chem. 2017 Oct 20;292(42):17561-17575. doi: 10.1074/jbc.M117.790378. Epub 2017 Aug 25. J Biol Chem. 2017. PMID: 28842479 Free PMC article.
-
Sustained TNF-α stimulation leads to transcriptional memory that greatly enhances signal sensitivity and robustness.Elife. 2020 Nov 6;9:e61965. doi: 10.7554/eLife.61965. Elife. 2020. PMID: 33155547 Free PMC article.
-
The roles of ubiquitination in extrinsic cell death pathways and its implications for therapeutics.Biochem Pharmacol. 2019 Apr;162:21-40. doi: 10.1016/j.bcp.2018.11.012. Epub 2018 Nov 16. Biochem Pharmacol. 2019. PMID: 30452908 Review.
-
Tumor necrosis factor-alpha induces VCAM-1-mediated inflammation via c-Src-dependent transactivation of EGF receptors in human cardiac fibroblasts.J Biomed Sci. 2015 Jul 15;22(1):53. doi: 10.1186/s12929-015-0165-8. J Biomed Sci. 2015. PMID: 26173590 Free PMC article.
-
TNF and ROS Crosstalk in Inflammation.Trends Cell Biol. 2016 Apr;26(4):249-261. doi: 10.1016/j.tcb.2015.12.002. Epub 2016 Jan 12. Trends Cell Biol. 2016. PMID: 26791157 Review.
Cited by
-
Treat the "Untreatable" by a Photothermal Agent: Triggering Heat and Immunological Responses for Rabies Virus Inactivation.Adv Sci (Weinh). 2023 Jan;10(2):e2205461. doi: 10.1002/advs.202205461. Epub 2022 Nov 17. Adv Sci (Weinh). 2023. PMID: 36385484 Free PMC article.
-
Effects of different depth of anesthesia on perioperative inflammatory reaction and hospital outcomes in elderly patients undergoing laparoscopic radical gastrectomy.BMC Anesthesiol. 2022 Oct 25;22(1):328. doi: 10.1186/s12871-022-01854-8. BMC Anesthesiol. 2022. PMID: 36284289 Free PMC article. Clinical Trial.
-
Re-Examining the Role of TNF in MS Pathogenesis and Therapy.Cells. 2020 Oct 14;9(10):2290. doi: 10.3390/cells9102290. Cells. 2020. PMID: 33066433 Free PMC article. Review.
-
Exploring causal relationships between inflammatory cytokines and allergic rhinitis, chronic rhinosinusitis, and nasal polyps: a Mendelian randomization study.Front Immunol. 2023 Nov 10;14:1288517. doi: 10.3389/fimmu.2023.1288517. eCollection 2023. Front Immunol. 2023. PMID: 38022554 Free PMC article.
-
Exploring the potential of Scabiosa columbaria in Alzheimer's disease treatment: An in silico approach.J Taibah Univ Med Sci. 2024 Sep 19;19(5):947-960. doi: 10.1016/j.jtumed.2024.09.003. eCollection 2024 Oct. J Taibah Univ Med Sci. 2024. PMID: 39397872 Free PMC article.
References
-
- Feldmann M. Translating molecular insights in autoimmunity into effective therapy. Annu. Rev. Immunol. 2009;27:1–27. - PubMed
-
- Cerami A. The value of failure: the discovery of TNF and its natural inhibitor erythropoietin. J. Intern. Med. 2011;269:8–15. - PubMed
-
- Lee RA, Eisen DB. Treatment of hidradenitis suppurativa with biologic medications. J. Am. Acad. Dermatol. 2015;73:S82–88. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
