

CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field
- PMID: 26631602
- PMCID: PMC4712441
- DOI: 10.1021/acs.jctc.5b00935
CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field
Abstract
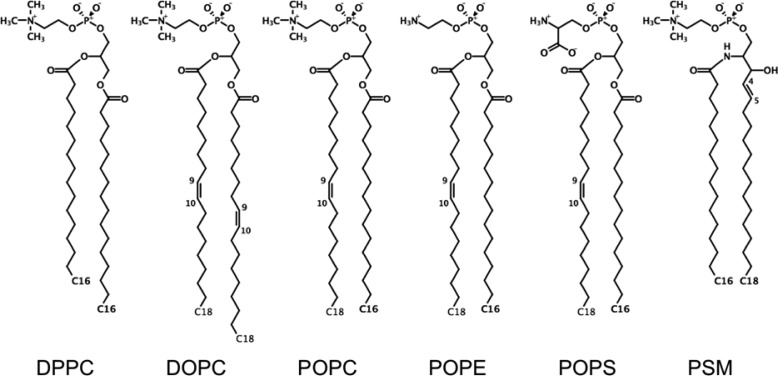
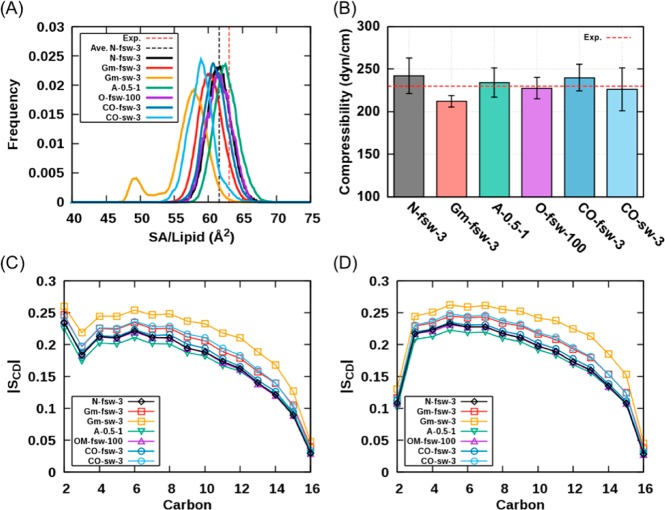
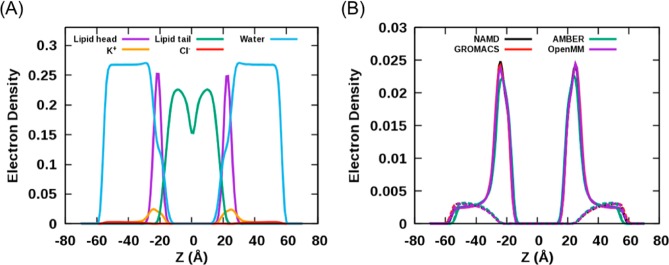
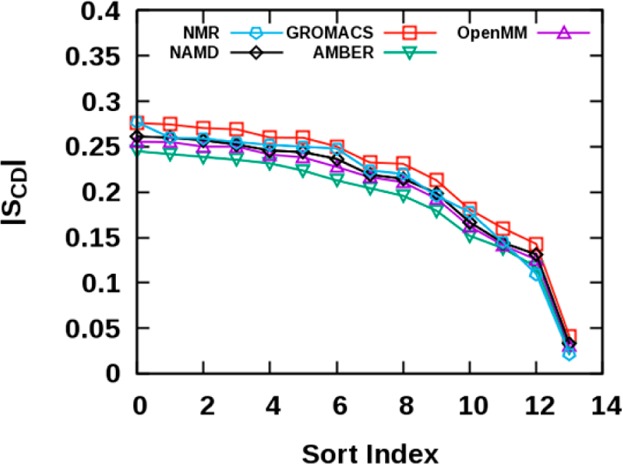
Proper treatment of nonbonded interactions is essential for the accuracy of molecular dynamics (MD) simulations, especially in studies of lipid bilayers. The use of the CHARMM36 force field (C36 FF) in different MD simulation programs can result in disagreements with published simulations performed with CHARMM due to differences in the protocols used to treat the long-range and 1-4 nonbonded interactions. In this study, we systematically test the use of the C36 lipid FF in NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM. A wide range of Lennard-Jones (LJ) cutoff schemes and integrator algorithms were tested to find the optimal simulation protocol to best match bilayer properties of six lipids with varying acyl chain saturation and head groups. MD simulations of a 1,2-dipalmitoyl-sn-phosphatidylcholine (DPPC) bilayer were used to obtain the optimal protocol for each program. MD simulations with all programs were found to reasonably match the DPPC bilayer properties (surface area per lipid, chain order parameters, and area compressibility modulus) obtained using the standard protocol used in CHARMM as well as from experiments. The optimal simulation protocol was then applied to the other five lipid simulations and resulted in excellent agreement between results from most simulation programs as well as with experimental data. AMBER compared least favorably with the expected membrane properties, which appears to be due to its use of the hard-truncation in the LJ potential versus a force-based switching function used to smooth the LJ potential as it approaches the cutoff distance. The optimal simulation protocol for each program has been implemented in CHARMM-GUI. This protocol is expected to be applicable to the remainder of the additive C36 FF including the proteins, nucleic acids, carbohydrates, and small molecules.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types.J Phys Chem B. 2010 Jun 17;114(23):7830-43. doi: 10.1021/jp101759q. J Phys Chem B. 2010. PMID: 20496934 Free PMC article.
-
Improving the CHARMM force field for polyunsaturated fatty acid chains.J Phys Chem B. 2012 Aug 9;116(31):9424-31. doi: 10.1021/jp304056p. Epub 2012 Jul 3. J Phys Chem B. 2012. PMID: 22697583
-
A systematic molecular dynamics simulation study of temperature dependent bilayer structural properties.Biochim Biophys Acta. 2014 Oct;1838(10):2520-9. doi: 10.1016/j.bbamem.2014.06.010. Epub 2014 Jun 19. Biochim Biophys Acta. 2014. PMID: 24953542
-
CHARMM-GUI 10 years for biomolecular modeling and simulation.J Comput Chem. 2017 Jun 5;38(15):1114-1124. doi: 10.1002/jcc.24660. Epub 2016 Nov 14. J Comput Chem. 2017. PMID: 27862047 Free PMC article. Review.
-
Mechanical properties of lipid bilayers from molecular dynamics simulation.Chem Phys Lipids. 2015 Nov;192:60-74. doi: 10.1016/j.chemphyslip.2015.07.014. Epub 2015 Jul 31. Chem Phys Lipids. 2015. PMID: 26238099 Free PMC article. Review.
Cited by
-
MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss.Exp Mol Med. 2024 Nov;56(11):2423-2435. doi: 10.1038/s12276-024-01338-4. Epub 2024 Nov 1. Exp Mol Med. 2024. PMID: 39482536 Free PMC article.
-
Network Pharmacology, Molecular Dynamics and In Vitro Assessments of Indigenous Herbal Formulations for Alzheimer's Therapy.Life (Basel). 2024 Sep 25;14(10):1222. doi: 10.3390/life14101222. Life (Basel). 2024. PMID: 39459522 Free PMC article.
-
PD-1 Targeted Antibody Discovery Using AI Protein Diffusion.Technol Cancer Res Treat. 2024 Jan-Dec;23:15330338241275947. doi: 10.1177/15330338241275947. Technol Cancer Res Treat. 2024. PMID: 39228166 Free PMC article.
-
Insights into the Microscopic Structure of RNF4-SIM-SUMO Complexes from MD Simulations.Biophys J. 2020 Oct 20;119(8):1558-1567. doi: 10.1016/j.bpj.2020.09.003. Epub 2020 Sep 11. Biophys J. 2020. PMID: 32976759 Free PMC article.
-
Modulation of Light Energy Transfer from Chromophore to Protein in the Channelrhodopsin ReaChR.Biophys J. 2020 Aug 4;119(3):705-716. doi: 10.1016/j.bpj.2020.06.031. Epub 2020 Jul 10. Biophys J. 2020. PMID: 32697975 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
