

CK2 acts as a potent negative regulator of receptor-mediated insulin release in vitro and in vivo
- PMID: 26598688
- PMCID: PMC4679045
- DOI: 10.1073/pnas.1519430112
CK2 acts as a potent negative regulator of receptor-mediated insulin release in vitro and in vivo
Abstract
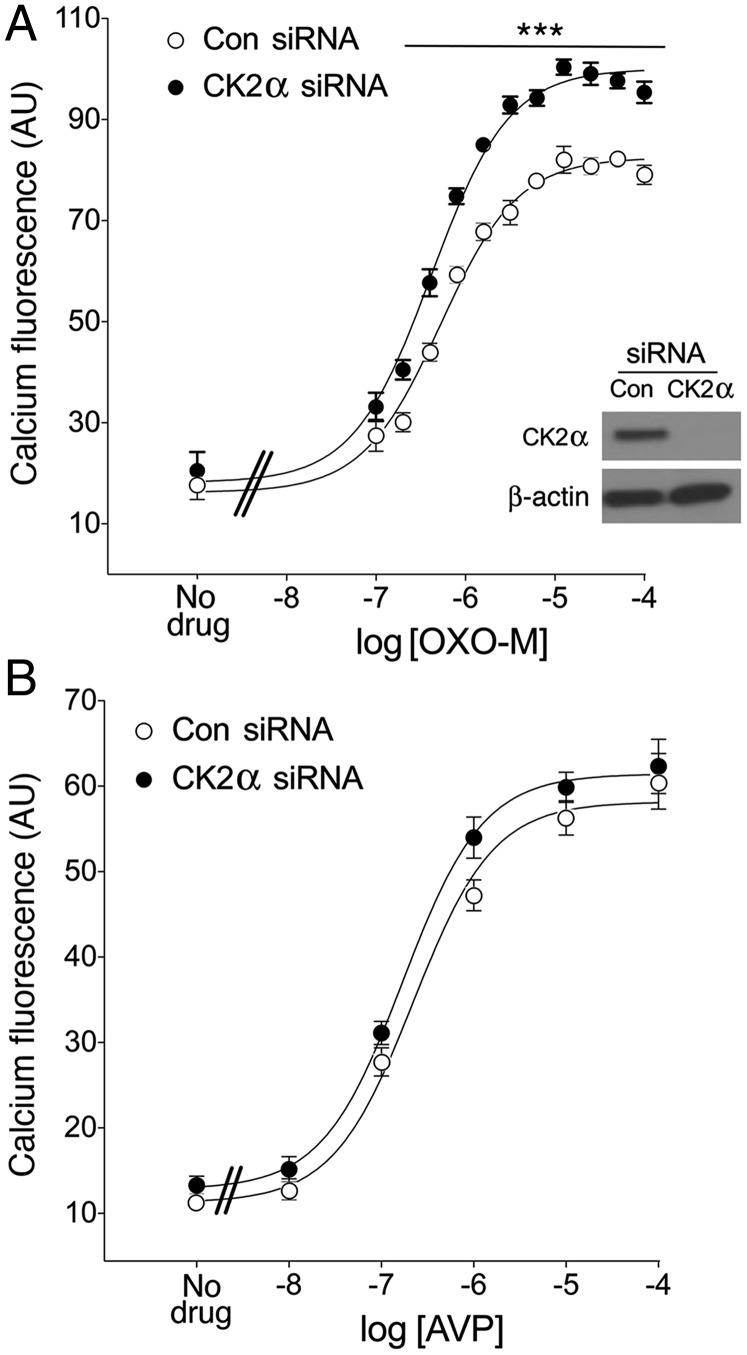
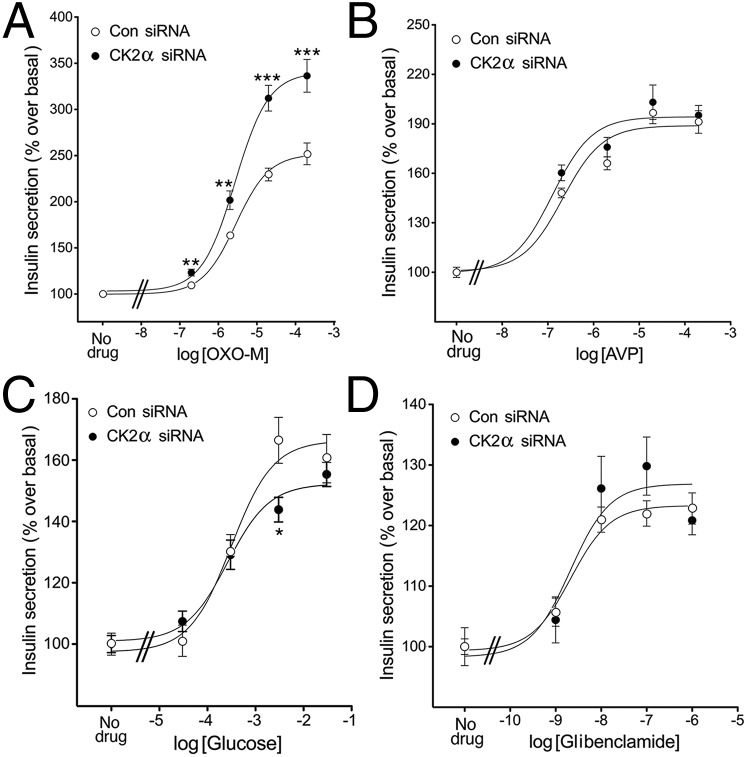
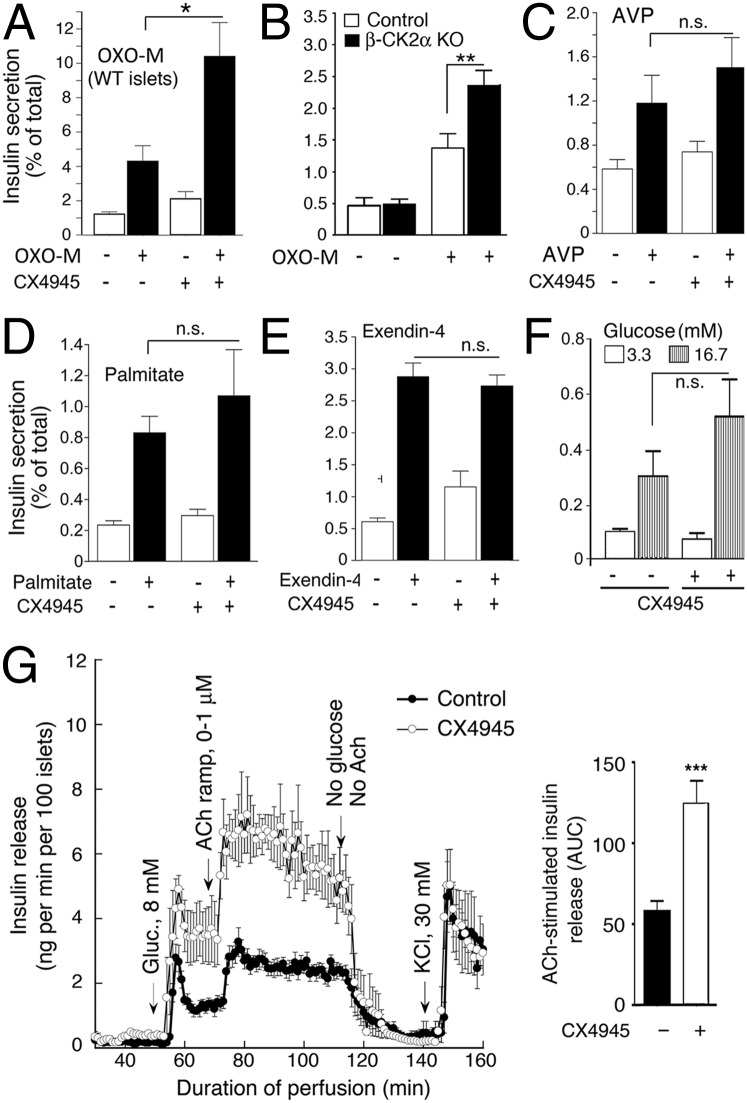
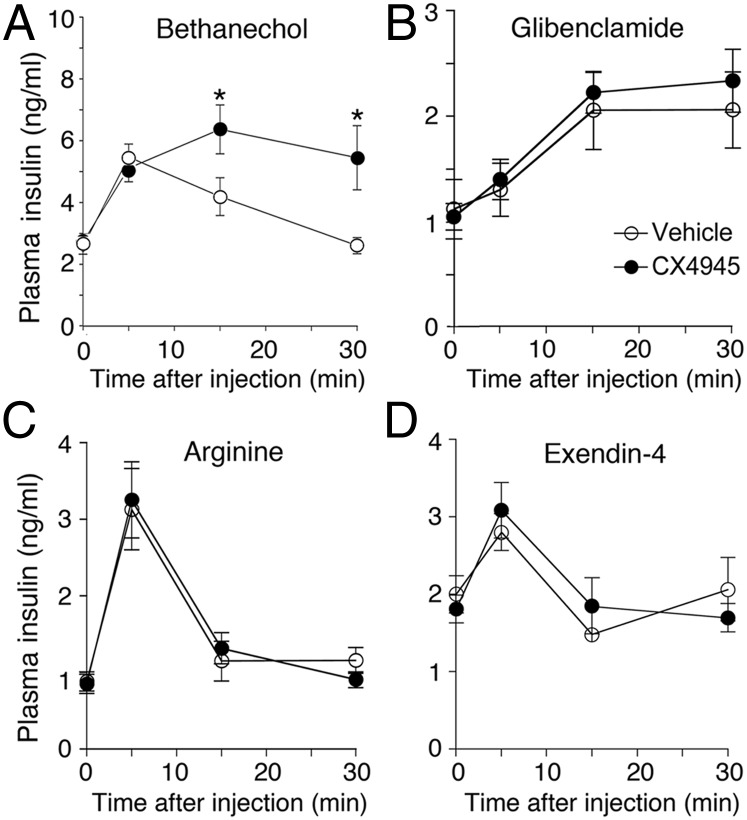
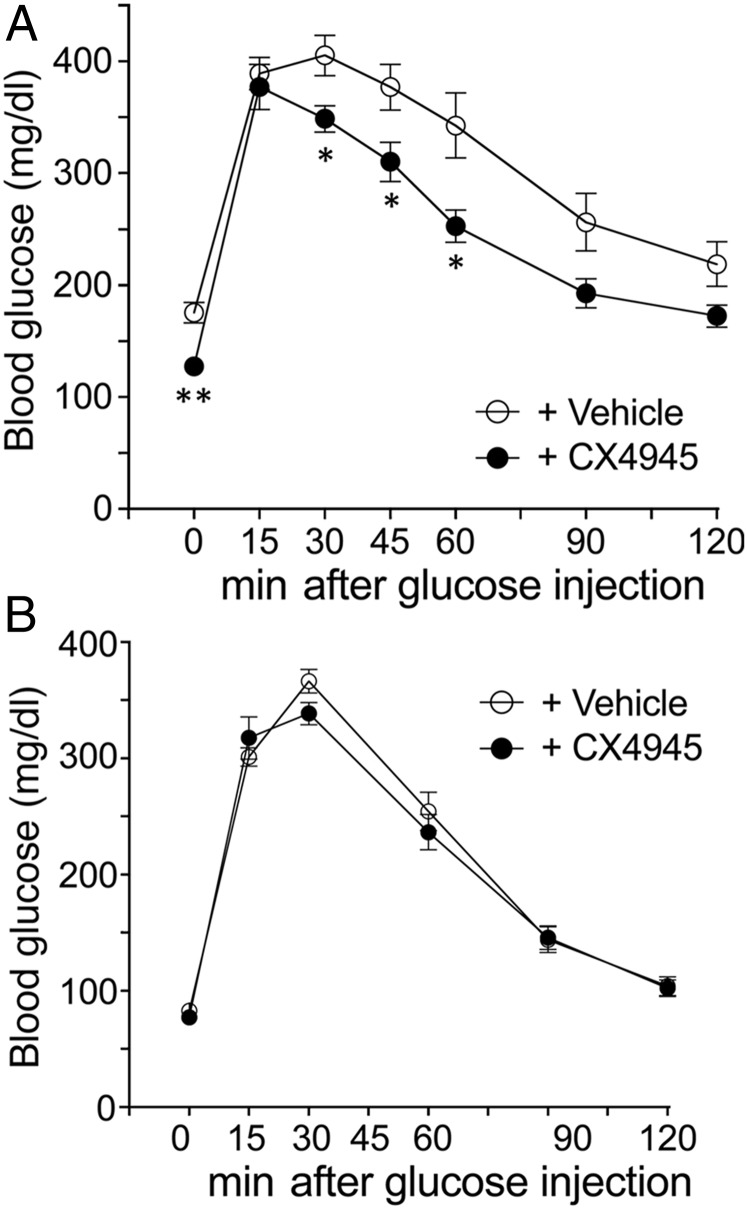
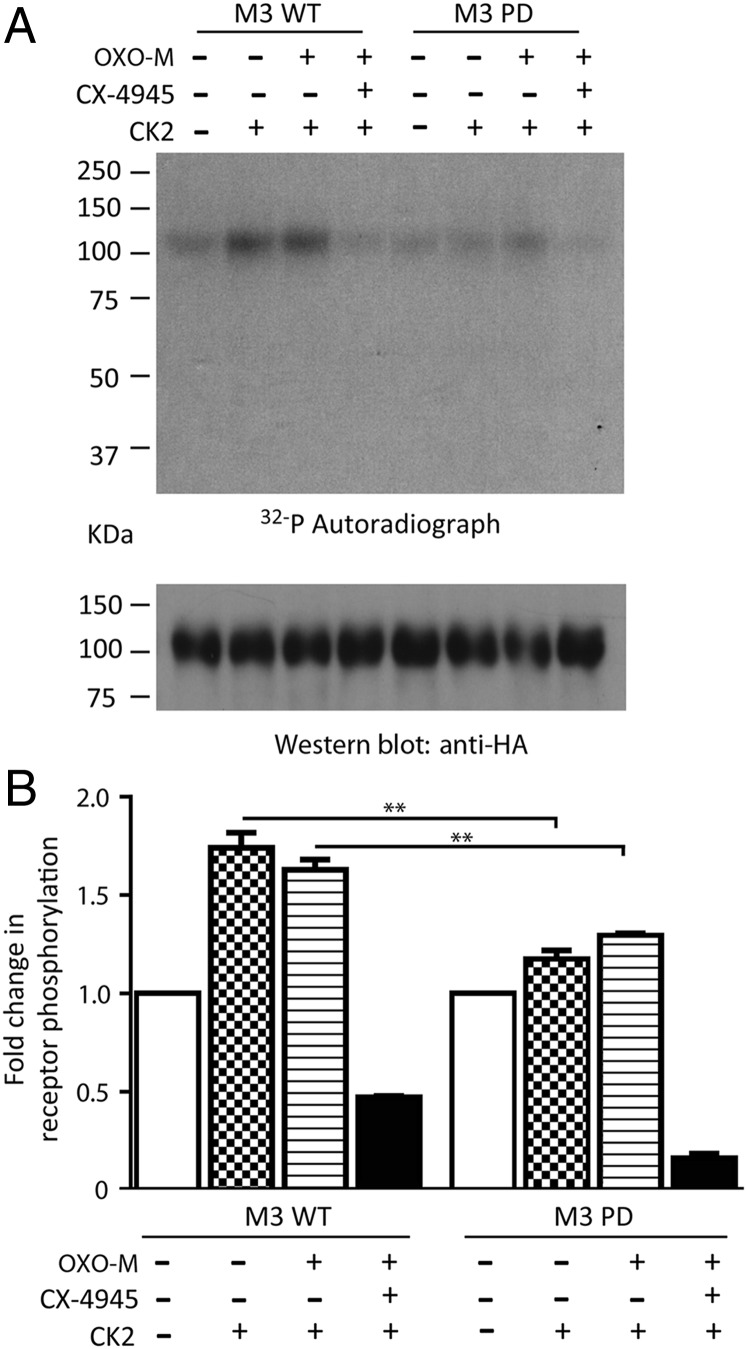
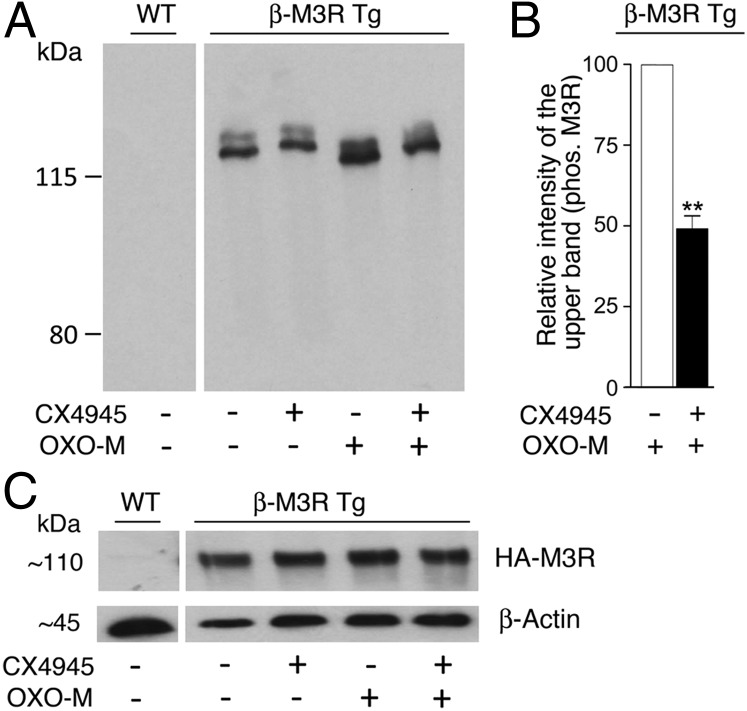
G protein-coupled receptors (GPCRs) regulate virtually all physiological functions including the release of insulin from pancreatic β-cells. β-Cell M3 muscarinic receptors (M3Rs) are known to play an essential role in facilitating insulin release and maintaining proper whole-body glucose homeostasis. As is the case with other GPCRs, M3R activity is regulated by phosphorylation by various kinases, including GPCR kinases and casein kinase 2 (CK2). At present, it remains unknown which of these various kinases are physiologically relevant for the regulation of β-cell activity. In the present study, we demonstrate that inhibition of CK2 in pancreatic β-cells, knockdown of CK2α expression, or genetic deletion of CK2α in β-cells of mutant mice selectively augmented M3R-stimulated insulin release in vitro and in vivo. In vitro studies showed that this effect was associated with an M3R-mediated increase in intracellular calcium levels. Treatment of mouse pancreatic islets with CX4945, a highly selective CK2 inhibitor, greatly reduced agonist-induced phosphorylation of β-cell M3Rs, indicative of CK2-mediated M3R phosphorylation. We also showed that inhibition of CK2 greatly enhanced M3R-stimulated insulin secretion in human islets. Finally, CX4945 treatment protected mice against diet-induced hyperglycemia and glucose intolerance in an M3R-dependent fashion. Our data demonstrate, for the first time to our knowledge, the physiological relevance of CK2 phosphorylation of a GPCR and suggest the novel concept that kinases acting on β-cell GPCRs may represent novel therapeutic targets.
Keywords: G protein-coupled receptors; GPCR regulation; glucose homeostasis; mouse models; β-cell function.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
RGS4 is a negative regulator of insulin release from pancreatic beta-cells in vitro and in vivo.Proc Natl Acad Sci U S A. 2010 Apr 27;107(17):7999-8004. doi: 10.1073/pnas.1003655107. Epub 2010 Apr 12. Proc Natl Acad Sci U S A. 2010. PMID: 20385802 Free PMC article.
-
Allosteric modulation of β-cell M3 muscarinic acetylcholine receptors greatly improves glucose homeostasis in lean and obese mice.Proc Natl Acad Sci U S A. 2019 Sep 10;116(37):18684-18690. doi: 10.1073/pnas.1904943116. Epub 2019 Aug 26. Proc Natl Acad Sci U S A. 2019. PMID: 31451647 Free PMC article.
-
Beneficial metabolic effects caused by persistent activation of beta-cell M3 muscarinic acetylcholine receptors in transgenic mice.Endocrinology. 2010 Nov;151(11):5185-94. doi: 10.1210/en.2010-0519. Epub 2010 Sep 15. Endocrinology. 2010. PMID: 20843999 Free PMC article.
-
Minireview: Novel aspects of M3 muscarinic receptor signaling in pancreatic β-cells.Mol Endocrinol. 2013 Aug;27(8):1208-16. doi: 10.1210/me.2013-1084. Epub 2013 Jul 2. Mol Endocrinol. 2013. PMID: 23820900 Free PMC article. Review.
-
Novel insights into the function of β-cell M3 muscarinic acetylcholine receptors: therapeutic implications.Trends Endocrinol Metab. 2011 Feb;22(2):74-80. doi: 10.1016/j.tem.2010.10.004. Epub 2010 Nov 23. Trends Endocrinol Metab. 2011. PMID: 21106385 Free PMC article. Review.
Cited by
-
Inhibition of cholinergic potentiation of insulin secretion from pancreatic islets by chronic elevation of glucose and fatty acids: Protection by casein kinase 2 inhibitor.Mol Metab. 2017 Oct;6(10):1240-1253. doi: 10.1016/j.molmet.2017.07.017. Epub 2017 Aug 4. Mol Metab. 2017. PMID: 29031723 Free PMC article.
-
Contribution of parasympathetic muscarinic augmentation of insulin secretion to olanzapine-induced hyperinsulinemia.Am J Physiol Endocrinol Metab. 2018 Aug 1;315(2):E250-E257. doi: 10.1152/ajpendo.00315.2017. Epub 2017 Dec 19. Am J Physiol Endocrinol Metab. 2018. PMID: 29351487 Free PMC article. Clinical Trial.
-
CK2 activity is crucial for proper glucagon expression.Diabetologia. 2024 Jul;67(7):1368-1385. doi: 10.1007/s00125-024-06128-1. Epub 2024 Mar 20. Diabetologia. 2024. PMID: 38503901 Free PMC article.
-
GPCRs in Intracellular Compartments: New Targets for Drug Discovery.Biomolecules. 2022 Sep 22;12(10):1343. doi: 10.3390/biom12101343. Biomolecules. 2022. PMID: 36291552 Free PMC article. Review.
-
Effects of Casein Kinase 2 Alpha 1 Gene Expression on Mice Liver Susceptible to Type 2 Diabetes Mellitus and Obesity.Int J Med Sci. 2020 Jan 1;17(1):13-20. doi: 10.7150/ijms.37110. eCollection 2020. Int J Med Sci. 2020. PMID: 31929734 Free PMC article.
References
-
- Ahrén B. Islet G protein-coupled receptors as potential targets for treatment of type 2 diabetes. Nat Rev Drug Discov. 2009;8(5):369–385. - PubMed
-
- Amisten S, Salehi A, Rorsman P, Jones PM, Persaud SJ. An atlas and functional analysis of G-protein coupled receptors in human islets of Langerhans. Pharmacol Ther. 2013;139(3):359–391. - PubMed
-
- Ahrén B. Autonomic regulation of islet hormone secretion: Implications for health and disease. Diabetologia. 2000;43(4):393–410. - PubMed
-
- Gilon P, Henquin JC. Mechanisms and physiological significance of the cholinergic control of pancreatic β-cell function. Endocr Rev. 2001;22(5):565–604. - PubMed
-
- Duttaroy A, et al. Muscarinic stimulation of pancreatic insulin and glucagon release is abolished in m3 muscarinic acetylcholine receptor-deficient mice. Diabetes. 2004;53(7):1714–1720. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
