

microRNA-33 Regulates ApoE Lipidation and Amyloid-β Metabolism in the Brain
- PMID: 26538644
- PMCID: PMC4635126
- DOI: 10.1523/JNEUROSCI.2053-15.2015
microRNA-33 Regulates ApoE Lipidation and Amyloid-β Metabolism in the Brain
Abstract
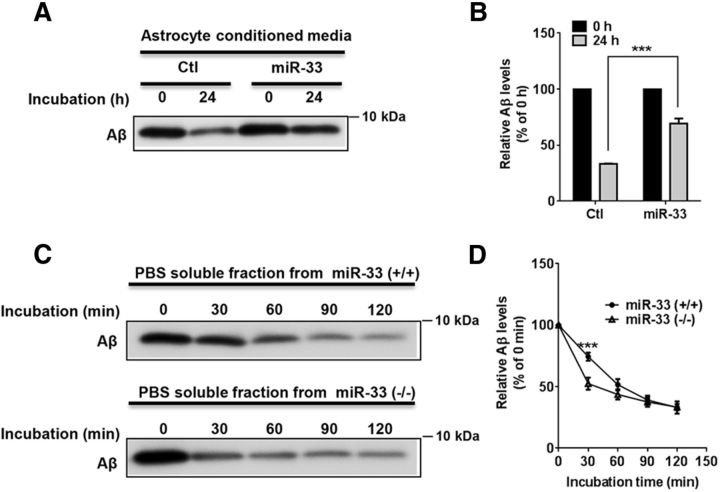
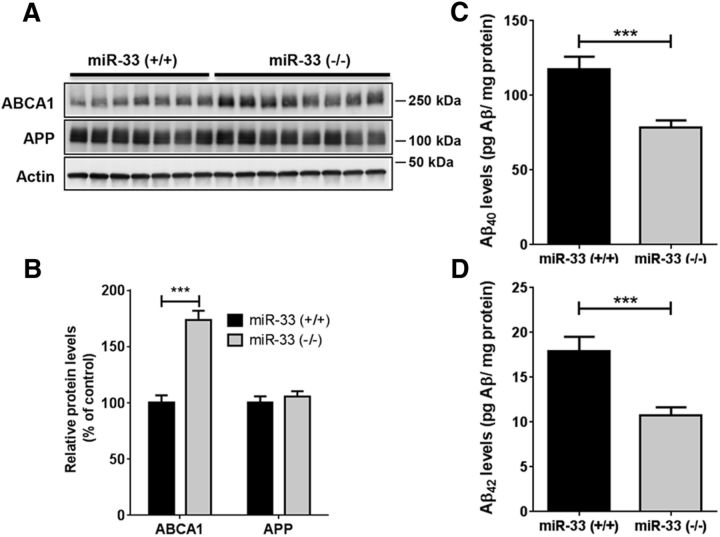
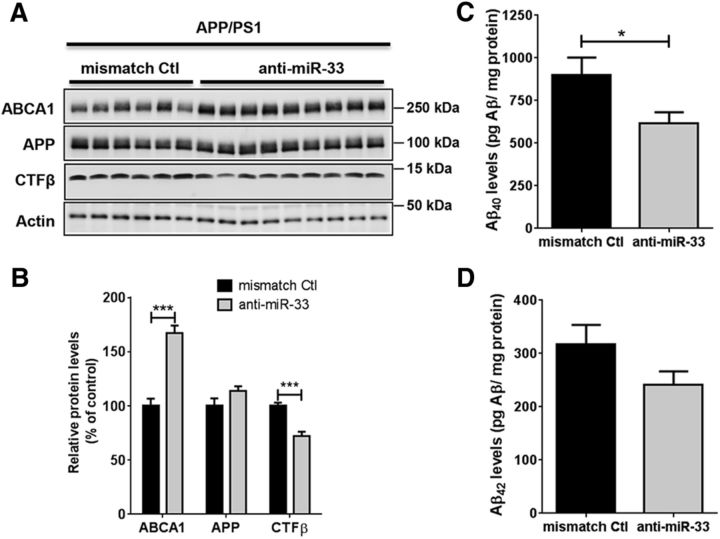
Dysregulation of amyloid-β (Aβ) metabolism is critical for Alzheimer's disease (AD) pathogenesis. Mounting evidence suggests that apolipoprotein E (ApoE) is involved in Aβ metabolism. ATP-binding cassette transporter A1 (ABCA1) is a key regulator of ApoE lipidation, which affects Aβ levels. Therefore, identifying regulatory mechanisms of ABCA1 expression in the brain may provide new therapeutic targets for AD. Here, we demonstrate that microRNA-33 (miR-33) regulates ABCA1 and Aβ levels in the brain. Overexpression of miR-33 impaired cellular cholesterol efflux and dramatically increased extracellular Aβ levels by promoting Aβ secretion and impairing Aβ clearance in neural cells. In contrast, genetic deletion of mir-33 in mice dramatically increased ABCA1 levels and ApoE lipidation, but it decreased endogenous Aβ levels in cortex. Most importantly, pharmacological inhibition of miR-33 via antisense oligonucleotide specifically in the brain markedly decreased Aβ levels in cortex of APP/PS1 mice, representing a potential therapeutic strategy for AD.
Significance statement: Brain lipid metabolism, in particular Apolipoprotein E (ApoE) lipidation, is critical to Aβ metabolism and Alzheimer's disease (AD). Brain lipid metabolism is largely separated from the periphery due to blood-brain barrier and different repertoire of lipoproteins. Therefore, identifying the novel regulatory mechanism of brain lipid metabolism may provide a new therapeutic strategy for AD. Although there have been studies on brain lipid metabolism, its regulation, in particular by microRNAs, is relatively unknown. Here, we demonstrate that inhibition of microRNA-33 increases lipidation of brain ApoE and reduces Aβ levels by inducing ABCA1. We provide a unique approach for AD therapeutics to increase ApoE lipidation and reduce Aβ levels via pharmacological inhibition of microRNA in vivo.
Keywords: ABCA1; Alzheimer's disease; ApoE; abeta; miR-33.
Copyright © 2015 the authors 0270-6474/15/3514718-10$15.00/0.
Figures

Comment in
-
Understanding the Role of miR-33 in Brain Lipid Metabolism: Implications for Alzheimer's Disease.J Neurosci. 2016 Mar 2;36(9):2558-60. doi: 10.1523/JNEUROSCI.4571-15.2016. J Neurosci. 2016. PMID: 26936997 Free PMC article. No abstract available.
Similar articles
-
Deletion of miR-33, a regulator of the ABCA1-APOE pathway, ameliorates neuropathological phenotypes in APP/PS1 mice.Alzheimers Dement. 2024 Nov;20(11):7805-7818. doi: 10.1002/alz.14243. Epub 2024 Sep 30. Alzheimers Dement. 2024. PMID: 39345217 Free PMC article.
-
A novel apoE-mimetic increases brain apoE levels, reduces Aβ pathology and improves memory when treated before onset of pathology in male mice that express APOE3.Alzheimers Res Ther. 2023 Dec 15;15(1):216. doi: 10.1186/s13195-023-01353-z. Alzheimers Res Ther. 2023. PMID: 38102668 Free PMC article.
-
ABCA1 is Necessary for Bexarotene-Mediated Clearance of Soluble Amyloid Beta from the Hippocampus of APP/PS1 Mice.J Neuroimmune Pharmacol. 2016 Mar;11(1):61-72. doi: 10.1007/s11481-015-9627-8. Epub 2015 Jul 15. J Neuroimmune Pharmacol. 2016. PMID: 26175148 Free PMC article.
-
Greasing the wheels of Abeta clearance in Alzheimer's disease: the role of lipids and apolipoprotein E.Biofactors. 2009 May-Jun;35(3):239-48. doi: 10.1002/biof.37. Biofactors. 2009. PMID: 19472365 Review.
-
ATP-binding cassette transporter A1: from metabolism to neurodegeneration.Neurobiol Dis. 2014 Dec;72 Pt A:13-21. doi: 10.1016/j.nbd.2014.05.007. Epub 2014 May 17. Neurobiol Dis. 2014. PMID: 24844148 Free PMC article. Review.
Cited by
-
MicroRNAs in Alzheimer's Disease: Function and Potential Applications as Diagnostic Biomarkers.Front Mol Neurosci. 2020 Aug 21;13:160. doi: 10.3389/fnmol.2020.00160. eCollection 2020. Front Mol Neurosci. 2020. PMID: 32973449 Free PMC article. Review.
-
Therapeutic approaches targeting Apolipoprotein E function in Alzheimer's disease.Mol Neurodegener. 2020 Jan 31;15(1):8. doi: 10.1186/s13024-020-0358-9. Mol Neurodegener. 2020. PMID: 32005122 Free PMC article. Review.
-
Apolipoprotein E metabolism and functions in brain and its role in Alzheimer's disease.Curr Opin Lipidol. 2017 Feb;28(1):60-67. doi: 10.1097/MOL.0000000000000383. Curr Opin Lipidol. 2017. PMID: 27922847 Free PMC article. Review.
-
A new perspective on microRNA-guided gene regulation specificity, and its potential generalization to transcription factors and RNA-binding proteins.Nucleic Acids Res. 2024 Sep 9;52(16):9360-9368. doi: 10.1093/nar/gkae694. Nucleic Acids Res. 2024. PMID: 39149906 Free PMC article. Review.
-
Deletion of miR-33, a regulator of the ABCA1-APOE pathway, ameliorates neuropathological phenotypes in APP/PS1 mice.Alzheimers Dement. 2024 Nov;20(11):7805-7818. doi: 10.1002/alz.14243. Epub 2024 Sep 30. Alzheimers Dement. 2024. PMID: 39345217 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL107953/HL/NHLBI NIH HHS/United States
- AG005681/AG/NIA NIH HHS/United States
- P50 AG016574/AG/NIA NIH HHS/United States
- R01 AG013956/AG/NIA NIH HHS/United States
- AG039708/AG/NIA NIH HHS/United States
- NS069329/NS/NINDS NIH HHS/United States
- R01 AG042513/AG/NIA NIH HHS/United States
- P50 AG005681/AG/NIA NIH HHS/United States
- AG016574/AG/NIA NIH HHS/United States
- R03 AG039708/AG/NIA NIH HHS/United States
- AG13956/AG/NIA NIH HHS/United States
- R37 AG013956/AG/NIA NIH HHS/United States
- P30 NS069329/NS/NINDS NIH HHS/United States
- AG042513/AG/NIA NIH HHS/United States
- HL107953/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous