

doi: 10.1186/s13059-015-0805-z.
ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis
Affiliations
- PMID: 26527291
- PMCID: PMC4630968
- DOI: 10.1186/s13059-015-0805-z
Item in Clipboard
ZIFA: Dimensionality reduction for zero-inflated single-cell gene expression analysis
Genome Biol.
.
Abstract
Single-cell RNA-seq data allows insight into normal cellular function and various disease states through molecular characterization of gene expression on the single cell level. Dimensionality reduction of such high-dimensional data sets is essential for visualization and analysis, but single-cell RNA-seq data are challenging for classical dimensionality-reduction methods because of the prevalence of dropout events, which lead to zero-inflated data. Here, we develop a dimensionality-reduction method, (Z)ero (I)nflated (F)actor (A)nalysis (ZIFA), which explicitly models the dropout characteristics, and show that it improves modeling accuracy on simulated and biological data sets.
Figures
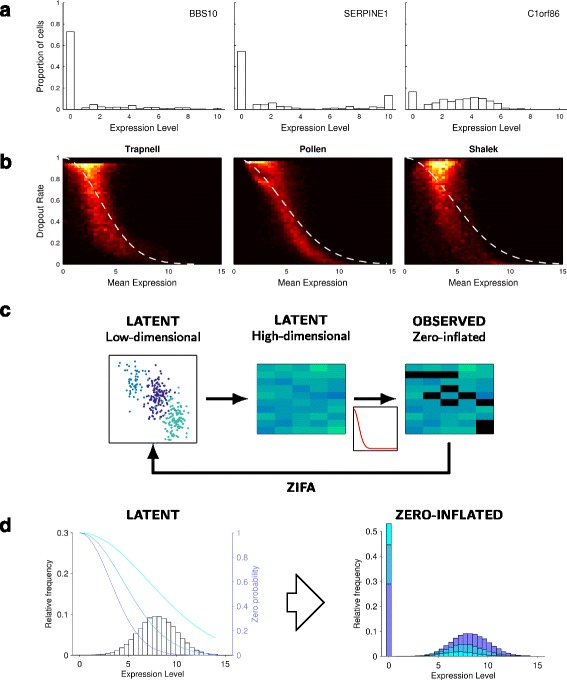
Zero-inflation in single-cell expression data. a Illustrative distribution of expression levels for three randomly chosen genes showing an abundance of single cells exhibiting null expression [15]. b Heat maps showing the relationship between dropout rate and mean non-zero expression level for three published single-cell data sets [3, 5, 14] including an approximate double exponential model fit. c Flow diagram illustrating the data generative process used by ZIFA. d Illustrative plot showing how different values of λ in the dropout-mean expression relationship (blue lines) can modulate the latent gene expression distribution to give a range of observed zero-inflated data
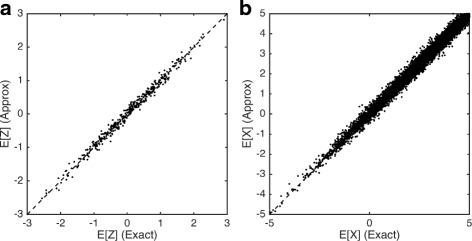
Comparison of exact and block-based EM algorithms. Plots show the correlation between expectations computed using the exact and block-based EM algorithms for latent low-dimensional positions (Z) (a) and latent observations X (b). Simulations were performed on a simulated data set with 500 genes and 200 cells. A block size of 50 was chosen for the approximate approach
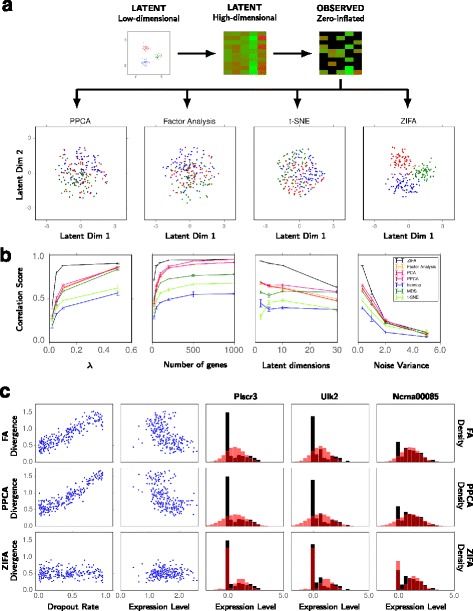
Performance comparison of dimensionality-reduction techniques. a Toy simulated data example illustrating the performance of ZIFA compared to standard dimensionality-reduction algorithms. b Performance on simulated data sets based on correlation score between the estimated and true latent distances as a function of λ (larger λ, lower dropout rate), number of genes and latent dimensions, and noise level used in the simulations. c Plots showing the divergence between the predictive and empirical data distributions as a function of dropout rate and mean expression level for FA, PPCA and ZIFA. Illustrative predictive performance and model fits (red, color online) on the T-cell single-cell data set (black) [3]
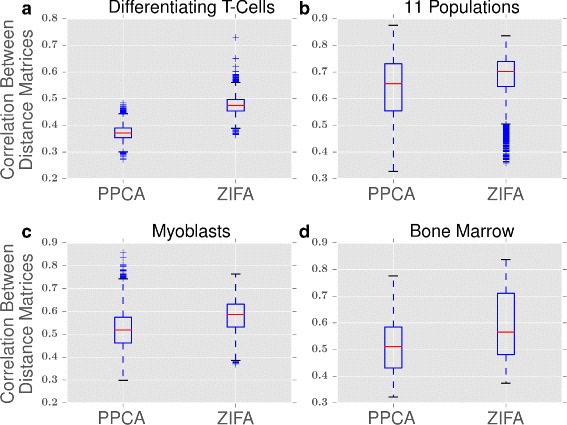
Consistency of cell-to-cell distances. Box plots showing the correlation between distance matrices for PPCA and ZIFA from 100 gene sets selected at random from (a) differentiating T cells [3], (b) 11 populations [15], (c) myoblasts [5] and (d) bone marrow [14]. The distance matrices produced by ZIFA are more correlated with each other than are the distance matrices produced by PPCA
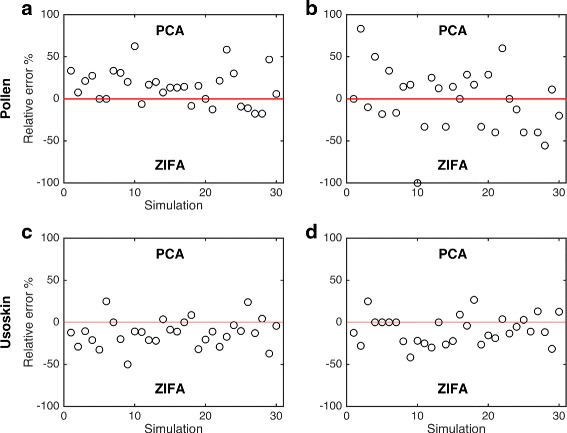
Cell type separability. Plot shows relative cell type misclassification error rates after applying PCA and ZIFA on random subset of 500 genes sampled for the Pollen [15] and Usoskin [16] data sets. Performance was measured based on error rates from (a, c) linear and (b, d) quadratic discriminant classifiers. Positive values indicate better performance based on PCA, and negative values for ZIFA
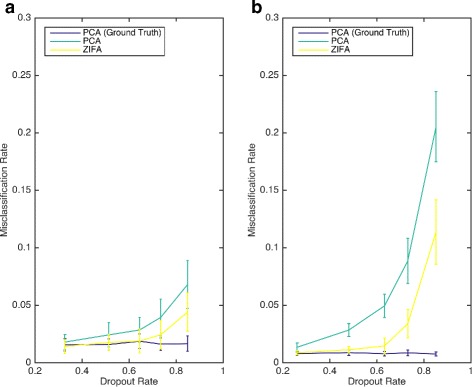
Understanding the relationship between cell type separability and dropout rate. This is a comparison of dimensionality-reduction techniques for cell typing. These plots show cell type misclassification rates (using QDA) as a function of dropout rate for the preprocessing using PCA and ZIFA on simulated data sets based on the (a) Pollen [15] and (b) Usoskin [16] data sets. The exact PCA results correspond to a ground-truth baseline when PCA is applied to simulated data with no dropout events
Similar articles
-
CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data.Genome Biol. 2017 Mar 28;18(1):59. doi: 10.1186/s13059-017-1188-0. Genome Biol. 2017. PMID: 28351406 Free PMC article.
-
Visualization of Single Cell RNA-Seq Data Using t-SNE in R.Methods Mol Biol. 2020;2117:159-167. doi: 10.1007/978-1-0716-0301-7_8. Methods Mol Biol. 2020. PMID: 31960377
-
FastProject: a tool for low-dimensional analysis of single-cell RNA-Seq data.BMC Bioinformatics. 2016 Aug 23;17(1):315. doi: 10.1186/s12859-016-1176-5. BMC Bioinformatics. 2016. PMID: 27553427 Free PMC article.
-
Bayesian gamma-negative binomial modeling of single-cell RNA sequencing data.BMC Genomics. 2020 Sep 9;21(Suppl 9):585. doi: 10.1186/s12864-020-06938-8. BMC Genomics. 2020. PMID: 32900358 Free PMC article.
-
Machine learning and statistical methods for clustering single-cell RNA-sequencing data.Brief Bioinform. 2020 Jul 15;21(4):1209-1223. doi: 10.1093/bib/bbz063. Brief Bioinform. 2020. PMID: 31243426 Review.
Cited by
-
Latent Factor Modeling of scRNA-Seq Data Uncovers Dysregulated Pathways in Autoimmune Disease Patients.iScience. 2020 Aug 12;23(9):101451. doi: 10.1016/j.isci.2020.101451. eCollection 2020 Sep 25. iScience. 2020. PMID: 32853994 Free PMC article.
-
Machine Intelligence in Single-Cell Data Analysis: Advances and New Challenges.Front Genet. 2021 May 31;12:655536. doi: 10.3389/fgene.2021.655536. eCollection 2021. Front Genet. 2021. PMID: 34135939 Free PMC article. Review.
-
SIMPLEs: a single-cell RNA sequencing imputation strategy preserving gene modules and cell clusters variation.NAR Genom Bioinform. 2020 Dec;2(4):lqaa077. doi: 10.1093/nargab/lqaa077. Epub 2020 Sep 28. NAR Genom Bioinform. 2020. PMID: 33029585 Free PMC article.
-
Coupling gene expression dynamics to cell size dynamics and cell cycle events: Exact and approximate solutions of the extended telegraph model.iScience. 2022 Dec 7;26(1):105746. doi: 10.1016/j.isci.2022.105746. eCollection 2023 Jan 20. iScience. 2022. PMID: 36619980 Free PMC article.
-
Differential Expression Analysis of Single-Cell RNA-Seq Data: Current Statistical Approaches and Outstanding Challenges.Entropy (Basel). 2022 Jul 18;24(7):995. doi: 10.3390/e24070995. Entropy (Basel). 2022. PMID: 35885218 Free PMC article. Review.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
