

Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping
- PMID: 26492141
- PMCID: PMC4917295
- DOI: 10.1038/nprot.2015.122
Whole-body tissue stabilization and selective extractions via tissue-hydrogel hybrids for high-resolution intact circuit mapping and phenotyping
Abstract
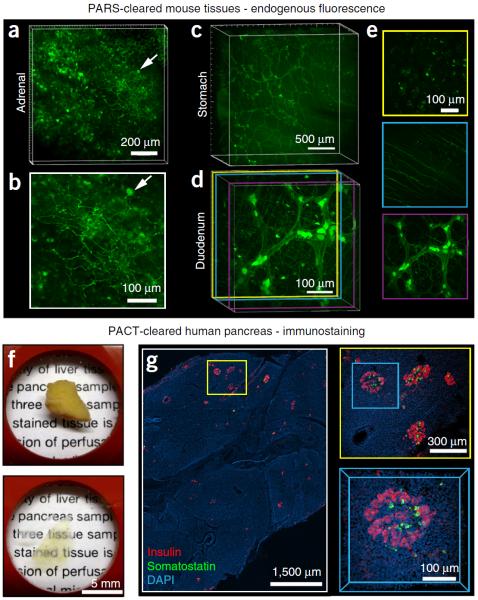
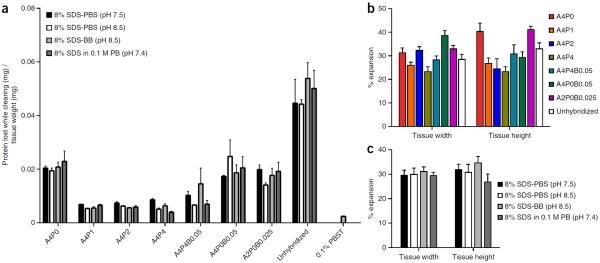
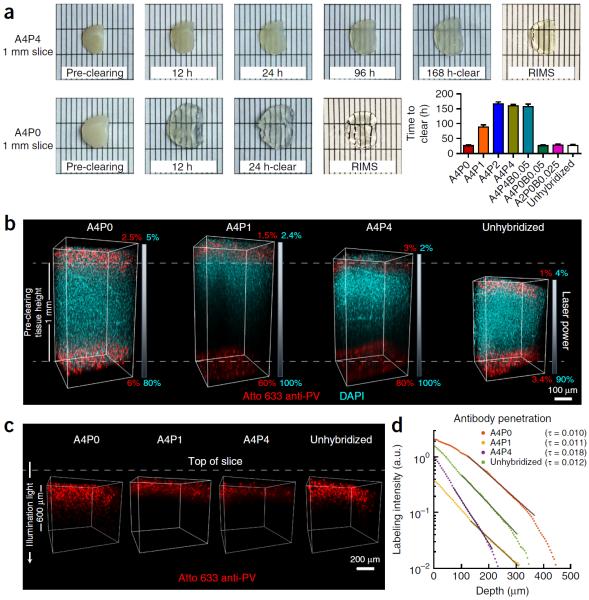
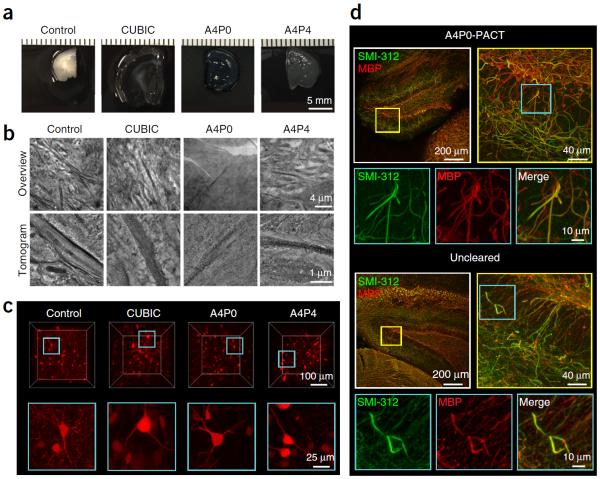
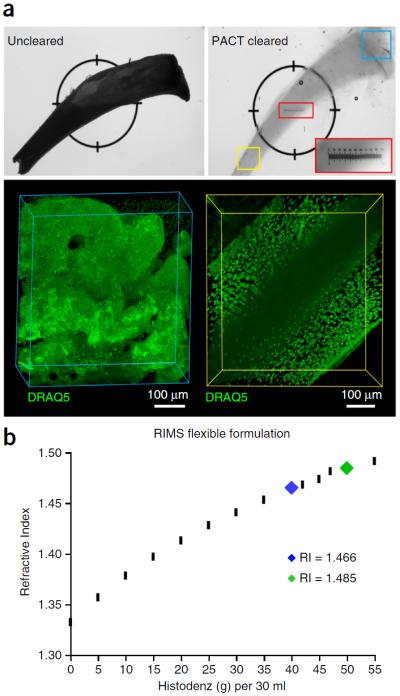
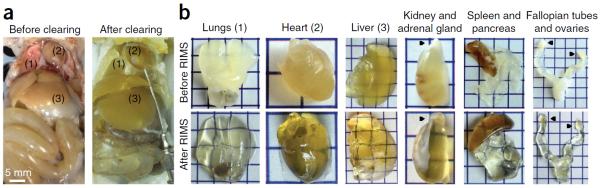
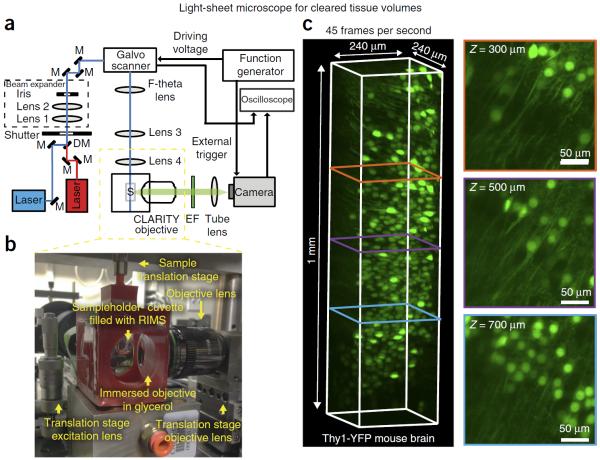
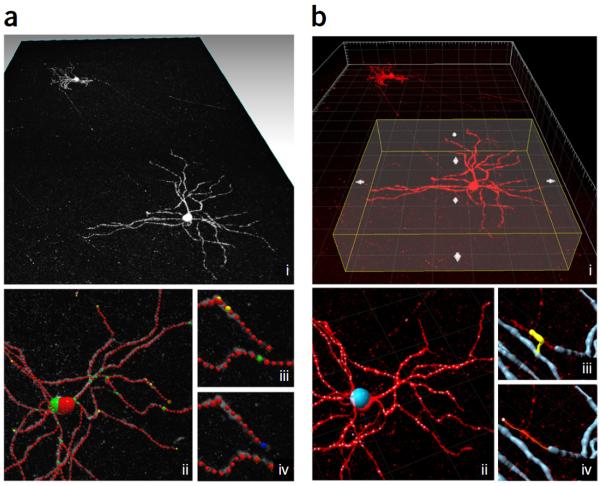
To facilitate fine-scale phenotyping of whole specimens, we describe here a set of tissue fixation-embedding, detergent-clearing and staining protocols that can be used to transform excised organs and whole organisms into optically transparent samples within 1-2 weeks without compromising their cellular architecture or endogenous fluorescence. PACT (passive CLARITY technique) and PARS (perfusion-assisted agent release in situ) use tissue-hydrogel hybrids to stabilize tissue biomolecules during selective lipid extraction, resulting in enhanced clearing efficiency and sample integrity. Furthermore, the macromolecule permeability of PACT- and PARS-processed tissue hybrids supports the diffusion of immunolabels throughout intact tissue, whereas RIMS (refractive index matching solution) grants high-resolution imaging at depth by further reducing light scattering in cleared and uncleared samples alike. These methods are adaptable to difficult-to-image tissues, such as bone (PACT-deCAL), and to magnified single-cell visualization (ePACT). Together, these protocols and solutions enable phenotyping of subcellular components and tracing cellular connectivity in intact biological networks.
Figures


Similar articles
-
Single-cell phenotyping within transparent intact tissue through whole-body clearing.Cell. 2014 Aug 14;158(4):945-958. doi: 10.1016/j.cell.2014.07.017. Epub 2014 Jul 31. Cell. 2014. PMID: 25088144 Free PMC article.
-
Optimization of the optical transparency of rodent tissues by modified PACT-based passive clearing.Exp Mol Med. 2016 Dec 2;48(12):e274. doi: 10.1038/emm.2016.105. Exp Mol Med. 2016. PMID: 27909337 Free PMC article.
-
Elevated-temperature-induced acceleration of PACT clearing process of mouse brain tissue.Sci Rep. 2017 Jan 31;7:38848. doi: 10.1038/srep38848. Sci Rep. 2017. PMID: 28139694 Free PMC article.
-
Lectins for histochemical demonstration of glycans.Histochem Cell Biol. 2011 Aug;136(2):117-30. doi: 10.1007/s00418-011-0848-5. Epub 2011 Jul 31. Histochem Cell Biol. 2011. PMID: 21805335 Review.
-
A survey of clearing techniques for 3D imaging of tissues with special reference to connective tissue.Prog Histochem Cytochem. 2016 Aug;51(2):9-23. doi: 10.1016/j.proghi.2016.04.001. Epub 2016 Apr 14. Prog Histochem Cytochem. 2016. PMID: 27142295 Review.
Cited by
-
Preparation and Characterization of Acrylic and Methacrylic Phospholipid-Mimetic Polymer Hydrogels and Their Applications in Optical Tissue Clearing.Polymers (Basel). 2024 Jan 15;16(2):241. doi: 10.3390/polym16020241. Polymers (Basel). 2024. PMID: 38257040 Free PMC article.
-
[Development in Tissue Clearing Technology and Its Application in Neurodegenerative Diseases].Sichuan Da Xue Xue Bao Yi Xue Ban. 2021 May;52(3):350-356. doi: 10.12182/20210560302. Sichuan Da Xue Xue Bao Yi Xue Ban. 2021. PMID: 34018350 Free PMC article. Review. Chinese.
-
Optimised tissue clearing minimises distortion and destruction during tissue delipidation.Neuropathol Appl Neurobiol. 2021 Apr;47(3):441-453. doi: 10.1111/nan.12673. Epub 2020 Dec 2. Neuropathol Appl Neurobiol. 2021. PMID: 33107057 Free PMC article.
-
ROCKETS - a novel one-for-all toolbox for light sheet microscopy in drug discovery.Front Immunol. 2023 Feb 7;14:1034032. doi: 10.3389/fimmu.2023.1034032. eCollection 2023. Front Immunol. 2023. PMID: 36845124 Free PMC article.
-
Regionally specific levels and patterns of keratin 8 expression in the mouse embryo visceral endoderm emerge upon anterior-posterior axis determination.Front Cell Dev Biol. 2022 Dec 1;10:1037041. doi: 10.3389/fcell.2022.1037041. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36531946 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- P50 GM082545/GM/NIGMS NIH HHS/United States
- T32 GM007616/GM/NIGMS NIH HHS/United States
- 2 P50 GM082545-06/GM/NIGMS NIH HHS/United States
- R21 MH103824/MH/NIMH NIH HHS/United States
- 2T32GM007616-36/GM/NIGMS NIH HHS/United States
- U01 NS090577/NS/NINDS NIH HHS/United States
- R01 AG047664/AG/NIA NIH HHS/United States
- 1U01NS090577/NS/NINDS NIH HHS/United States
- IDP20D017782-01/PHS HHS/United States
- 1R01AG047664-01/AG/NIA NIH HHS/United States
- 1R21MH103824-01/MH/NIMH NIH HHS/United States
- DP2 NS087949/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
