

A Novel C-Terminal CIB2 (Calcium and Integrin Binding Protein 2) Mutation Associated with Non-Syndromic Hearing Loss in a Hispanic Family
- PMID: 26426422
- PMCID: PMC4591343
- DOI: 10.1371/journal.pone.0133082
A Novel C-Terminal CIB2 (Calcium and Integrin Binding Protein 2) Mutation Associated with Non-Syndromic Hearing Loss in a Hispanic Family
Erratum in
-
Correction: A Novel C-Terminal CIB2 (Calcium and Integrin Binding Protein 2) Mutation Associated with Non-Syndromic Hearing Loss in a Hispanic Family.PLoS One. 2015 Oct 16;10(10):e0141259. doi: 10.1371/journal.pone.0141259. eCollection 2015. PLoS One. 2015. PMID: 26473954 Free PMC article. No abstract available.
Abstract
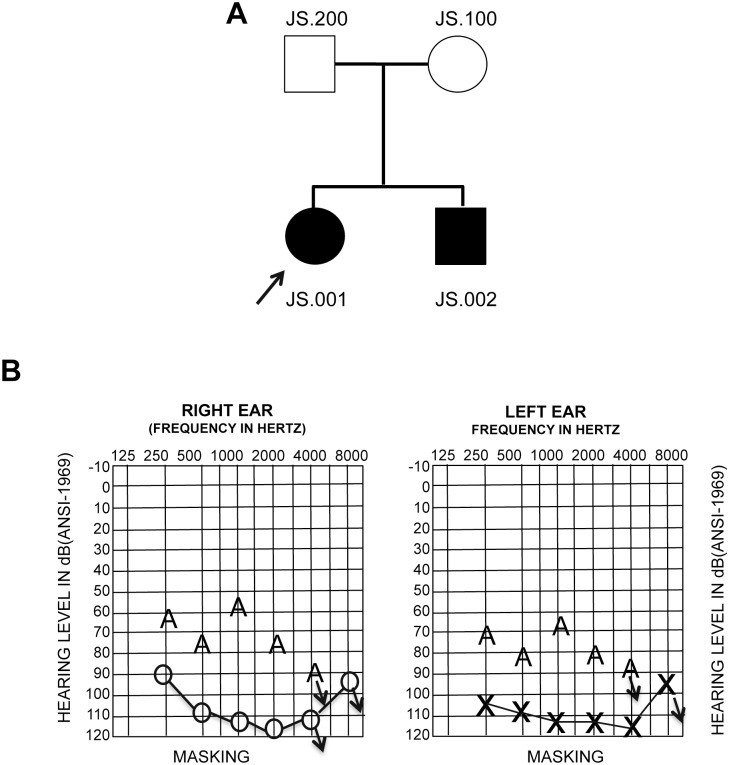
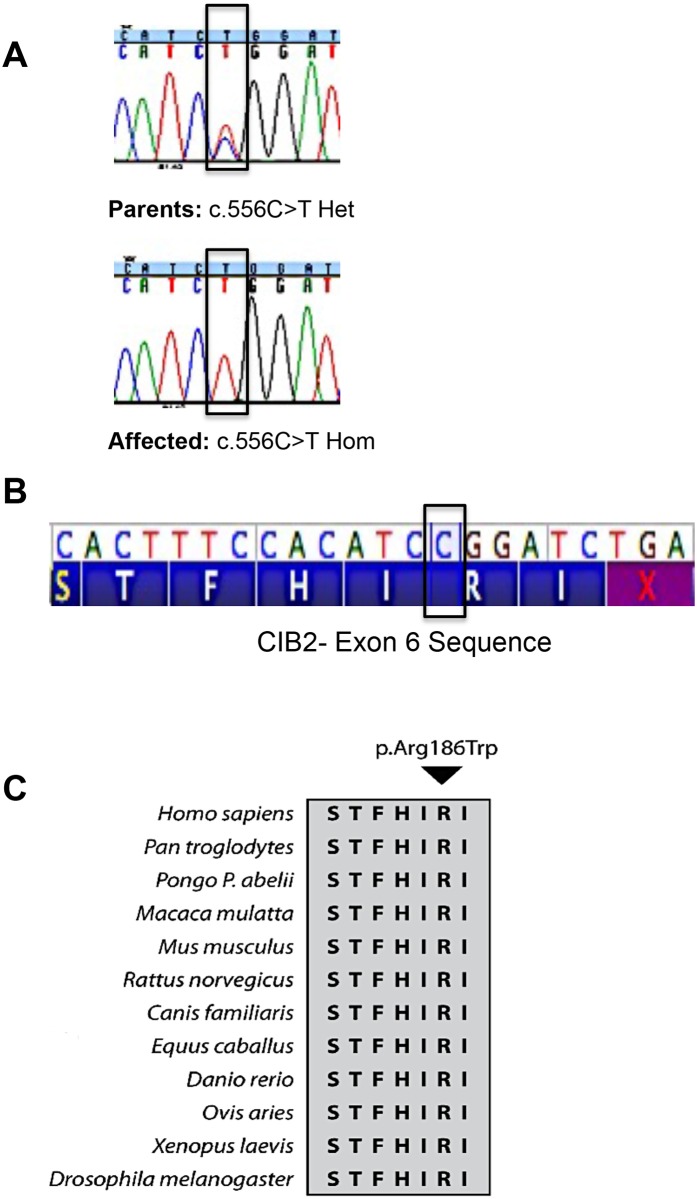
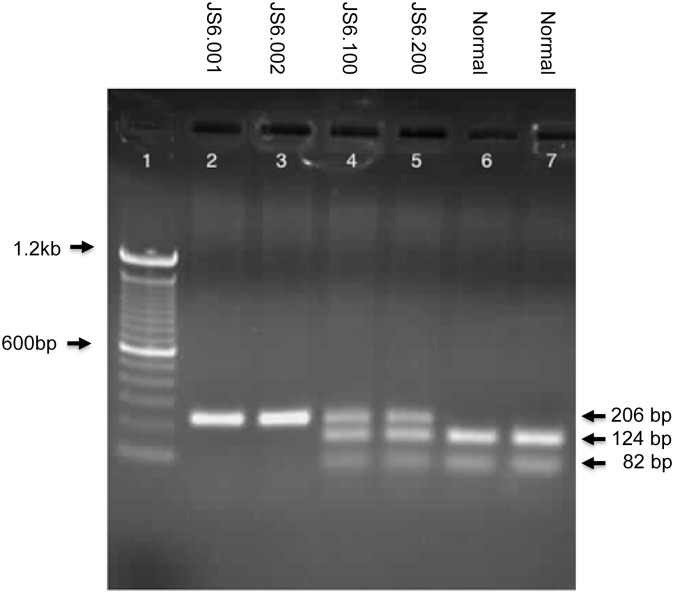
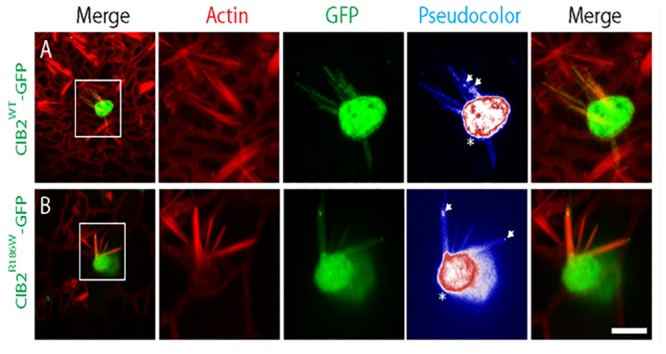
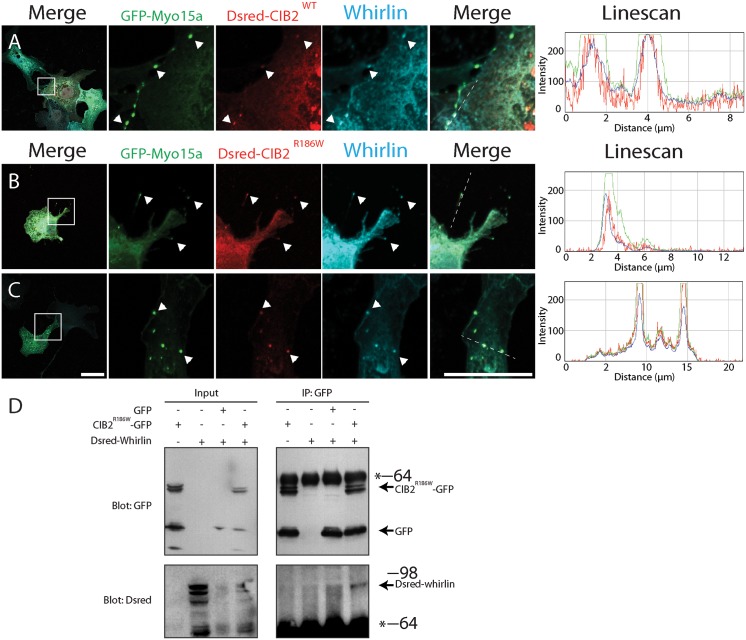
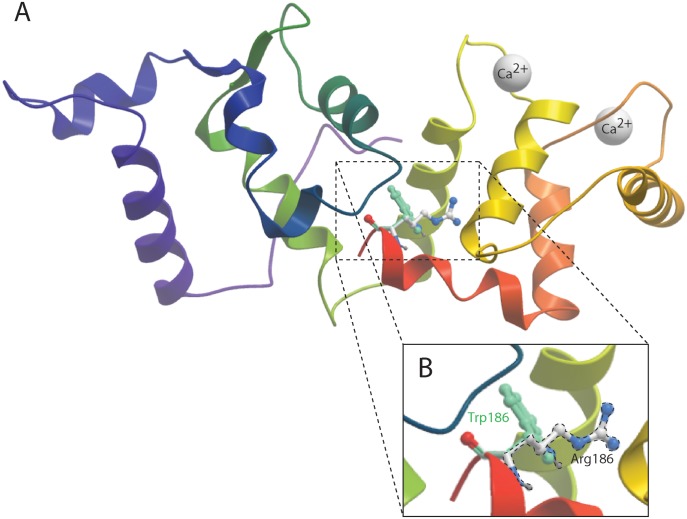
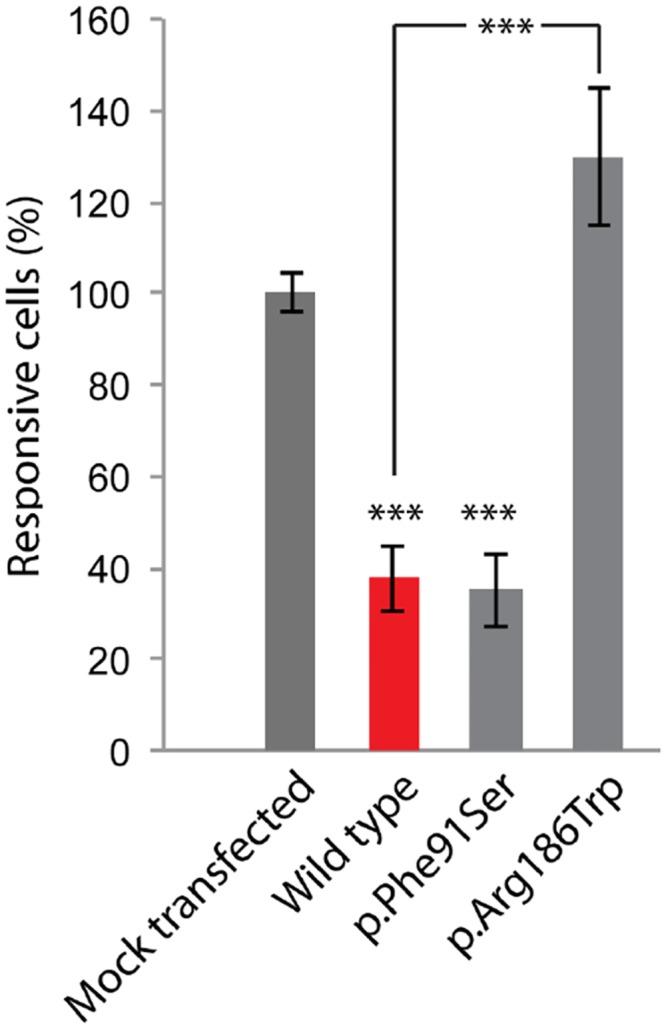
Hearing loss is a complex disorder caused by both genetic and environmental factors. Previously, mutations in CIB2 have been identified as a common cause of genetic hearing loss in Pakistani and Turkish populations. Here we report a novel (c.556C>T; p.(Arg186Trp)) transition mutation in the CIB2 gene identified through whole exome sequencing (WES) in a Caribbean Hispanic family with non-syndromic hearing loss. CIB2 belongs to the family of calcium-and integrin-binding (CIB) proteins. The carboxy-termini of CIB proteins are associated with calcium binding and intracellular signaling. The p.(Arg186Trp) mutation is localized within predicted type II PDZ binding ligand at the carboxy terminus. Our ex vivo studies revealed that the mutation did not alter the interactions of CIB2 with Whirlin, nor its targeting to the tips of hair cell stereocilia. However, we found that the mutation disrupts inhibition of ATP-induced Ca2+ responses by CIB2 in a heterologous expression system. Our findings support p.(Arg186Trp) mutation as a cause for hearing loss in this Hispanic family. In addition, it further highlights the necessity of the calcium binding property of CIB2 for normal hearing.
Conflict of interest statement
Figures
Similar articles
-
Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides.Int J Mol Sci. 2022 Mar 24;23(7):3552. doi: 10.3390/ijms23073552. Int J Mol Sci. 2022. PMID: 35408910 Free PMC article. Review.
-
Novel and recurrent CIB2 variants, associated with nonsyndromic deafness, do not affect calcium buffering and localization in hair cells.Eur J Hum Genet. 2016 Apr;24(4):542-9. doi: 10.1038/ejhg.2015.157. Epub 2015 Jul 15. Eur J Hum Genet. 2016. PMID: 26173970 Free PMC article.
-
Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48.Nat Genet. 2012 Nov;44(11):1265-71. doi: 10.1038/ng.2426. Epub 2012 Sep 30. Nat Genet. 2012. PMID: 23023331 Free PMC article.
-
Advances in genetic hearing loss: CIB2 gene.Eur Arch Otorhinolaryngol. 2017 Apr;274(4):1791-1795. doi: 10.1007/s00405-016-4330-9. Epub 2016 Oct 22. Eur Arch Otorhinolaryngol. 2017. PMID: 27771768 Free PMC article. Review.
-
Characterization of a novel MYO3A missense mutation associated with a dominant form of late onset hearing loss.Sci Rep. 2018 Jun 7;8(1):8706. doi: 10.1038/s41598-018-26818-2. Sci Rep. 2018. PMID: 29880844 Free PMC article. Clinical Trial.
Cited by
-
Evolutionary-Conserved Allosteric Properties of Three Neuronal Calcium Sensor Proteins.Front Mol Neurosci. 2019 Mar 7;12:50. doi: 10.3389/fnmol.2019.00050. eCollection 2019. Front Mol Neurosci. 2019. PMID: 30899213 Free PMC article.
-
The genetic and phenotypic landscapes of Usher syndrome: from disease mechanisms to a new classification.Hum Genet. 2022 Apr;141(3-4):709-735. doi: 10.1007/s00439-022-02448-7. Epub 2022 Mar 30. Hum Genet. 2022. PMID: 35353227 Free PMC article. Review.
-
CRISPR/Cas9: targeted genome editing for the treatment of hereditary hearing loss.J Appl Genet. 2020 Feb;61(1):51-65. doi: 10.1007/s13353-019-00535-6. Epub 2020 Jan 7. J Appl Genet. 2020. PMID: 31912450 Review.
-
Associations Between TGFA/TGFB3/MSX1 Gene Polymorphisms and Congenital Non-Syndromic Hearing Impairment in a Chinese Population.Med Sci Monit. 2016 Jun 29;22:2253-66. doi: 10.12659/msm.896527. Med Sci Monit. 2016. PMID: 27356075 Free PMC article.
-
Calcium- and Integrin-Binding Protein 2 (CIB2) in Physiology and Disease: Bright and Dark Sides.Int J Mol Sci. 2022 Mar 24;23(7):3552. doi: 10.3390/ijms23073552. Int J Mol Sci. 2022. PMID: 35408910 Free PMC article. Review.
References
-
- Morton CC, Nance WE. Newborn hearing screening—a silent revolution. The New England journal of medicine. 2006;354(20):2151–64. . - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
