

An interconnected hierarchical model of cell death regulation by the BCL-2 family
- PMID: 26344567
- PMCID: PMC4589531
- DOI: 10.1038/ncb3236
An interconnected hierarchical model of cell death regulation by the BCL-2 family
Abstract
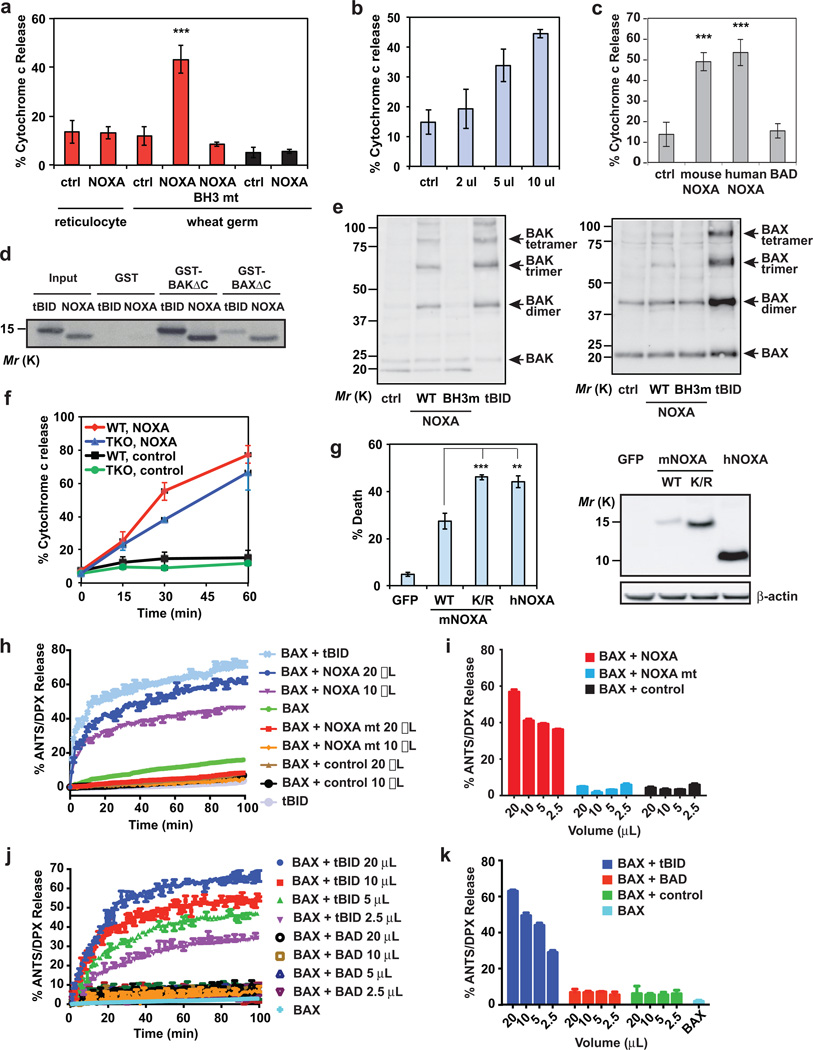
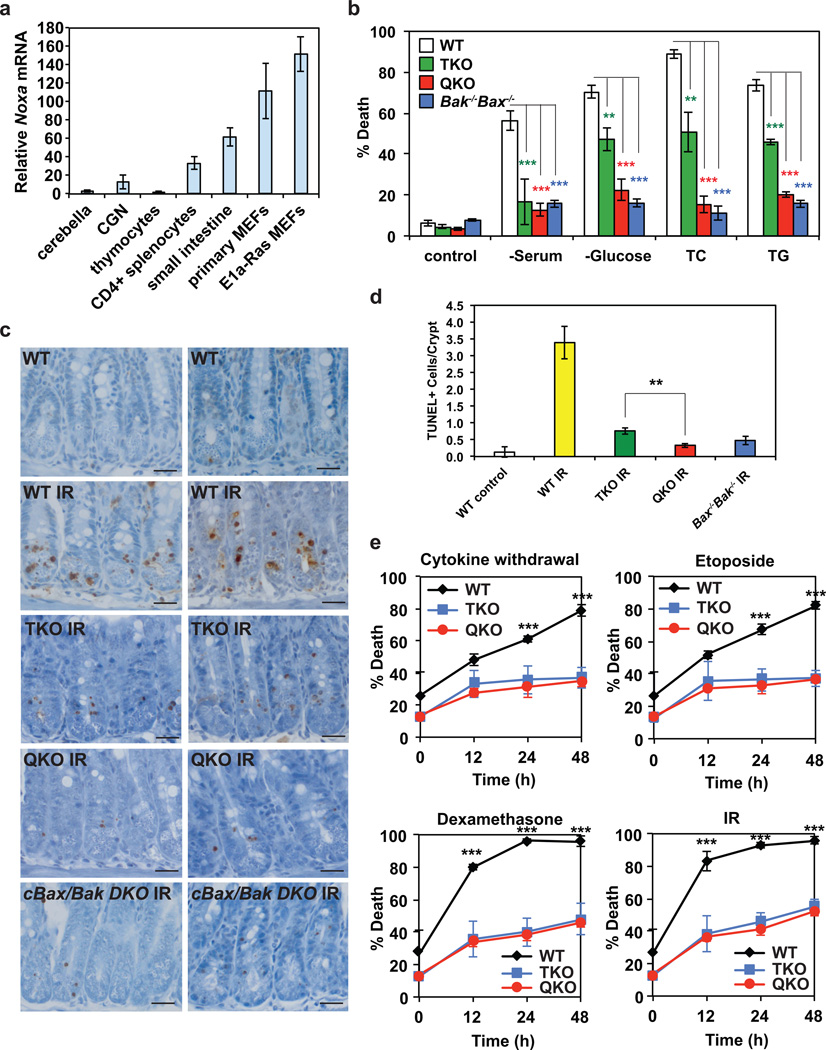
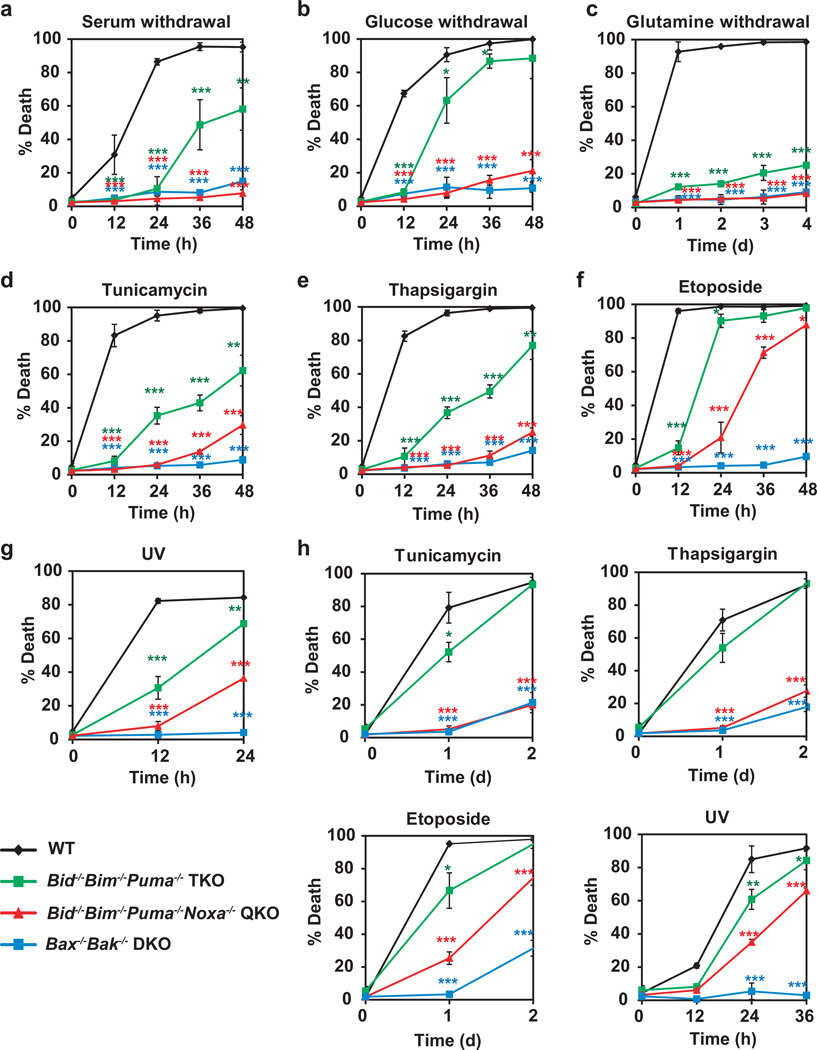
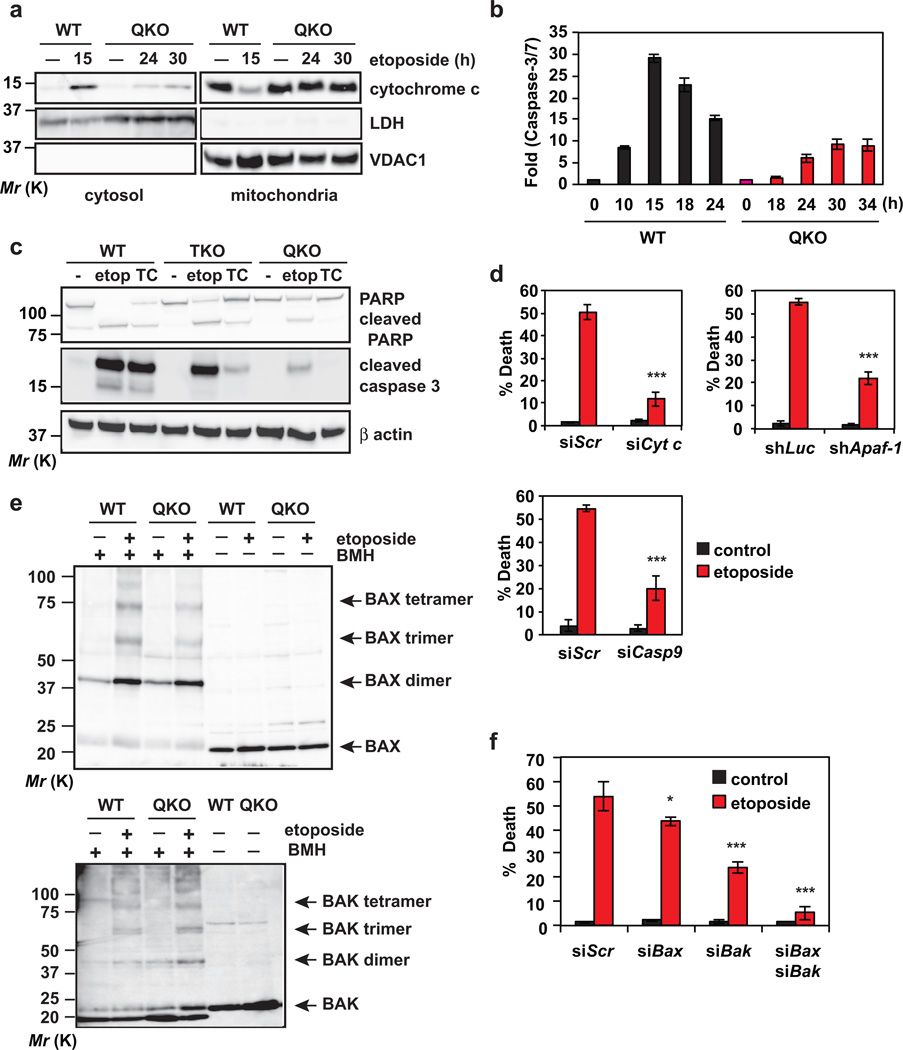
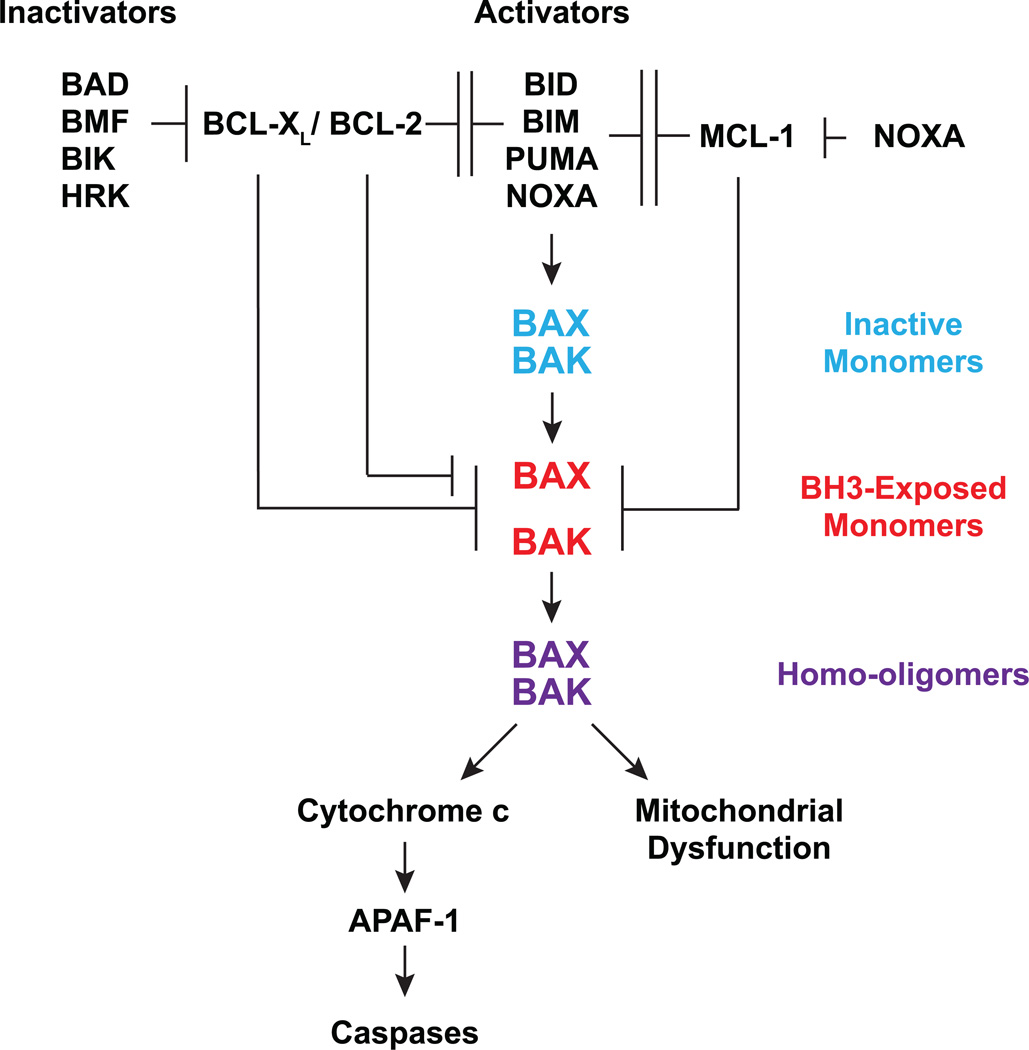
Multidomain pro-apoptotic BAX and BAK, once activated, permeabilize mitochondria to trigger apoptosis, whereas anti-apoptotic BCL-2 members preserve mitochondrial integrity. The BH3-only molecules (BH3s) promote apoptosis by either activating BAX-BAK or inactivating anti-apoptotic members. Here, we present biochemical and genetic evidence that NOXA is a bona fide activator BH3. Using combinatorial gain-of-function and loss-of-function approaches in Bid(-/-)Bim(-/-)Puma(-/-)Noxa(-/-) and Bax(-/-)Bak(-/-) cells, we have constructed an interconnected hierarchical model that accommodates and explains how the intricate interplays between the BCL-2 members dictate cellular survival versus death. BID, BIM, PUMA and NOXA directly induce stepwise, bimodal activation of BAX-BAK. BCL-2, BCL-XL and MCL-1 inhibit both modes of BAX-BAK activation by sequestering activator BH3s and 'BH3-exposed' monomers of BAX-BAK, respectively. Furthermore, autoactivation of BAX and BAK can occur independently of activator BH3s through downregulation of BCL-2, BCL-XL and MCL-1. Our studies lay a foundation for targeting the BCL-2 family for treating diseases with dysregulated apoptosis.
Figures
Similar articles
-
Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis.Mol Cell. 2009 Nov 13;36(3):487-99. doi: 10.1016/j.molcel.2009.09.030. Mol Cell. 2009. PMID: 19917256 Free PMC article.
-
The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner.J Biol Chem. 2011 Mar 18;286(11):9382-92. doi: 10.1074/jbc.M110.203638. Epub 2010 Dec 9. J Biol Chem. 2011. PMID: 21148306 Free PMC article.
-
Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies.Nat Cell Biol. 2006 Dec;8(12):1348-58. doi: 10.1038/ncb1499. Epub 2006 Nov 19. Nat Cell Biol. 2006. PMID: 17115033
-
BCL-2 proteins and apoptosis: Recent insights and unknowns.Biochem Biophys Res Commun. 2018 May 27;500(1):26-34. doi: 10.1016/j.bbrc.2017.06.190. Epub 2017 Jul 1. Biochem Biophys Res Commun. 2018. PMID: 28676391 Review.
-
Mitochondrial membrane permeabilisation by Bax/Bak.Biochem Biophys Res Commun. 2003 May 9;304(3):455-61. doi: 10.1016/s0006-291x(03)00617-x. Biochem Biophys Res Commun. 2003. PMID: 12729579 Review.
Cited by
-
Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis.J Lipid Res. 2016 Oct;57(10):1758-1770. doi: 10.1194/jlr.R066357. Epub 2016 Apr 5. J Lipid Res. 2016. PMID: 27049024 Free PMC article. Review.
-
BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis.Cell Res. 2019 Nov;29(11):942-952. doi: 10.1038/s41422-019-0231-y. Epub 2019 Sep 24. Cell Res. 2019. PMID: 31551537 Free PMC article.
-
SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes.Nat Cancer. 2022 Feb;3(2):188-202. doi: 10.1038/s43018-021-00316-3. Epub 2022 Feb 3. Nat Cancer. 2022. PMID: 35115713 Free PMC article.
-
The interplay between BAX and BAK tunes apoptotic pore growth to control mitochondrial-DNA-mediated inflammation.Mol Cell. 2022 Mar 3;82(5):933-949.e9. doi: 10.1016/j.molcel.2022.01.008. Epub 2022 Feb 3. Mol Cell. 2022. PMID: 35120587 Free PMC article.
-
Blockade of Cyclophilin D Attenuates Oxidative Stress-Induced Cell Death in Human Dental Pulp Cells.Oxid Med Cell Longev. 2019 Apr 4;2019:1729013. doi: 10.1155/2019/1729013. eCollection 2019. Oxid Med Cell Longev. 2019. PMID: 31089403 Free PMC article.
References
-
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. - PubMed
-
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. - PubMed
-
- Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. - PubMed
-
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. - PubMed
-
- Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
