

BRCA1 Is Required for Maintenance of Phospho-Chk1 and G2/M Arrest during DNA Cross-Link Repair in DT40 Cells
- PMID: 26324327
- PMCID: PMC4609749
- DOI: 10.1128/MCB.01497-14
BRCA1 Is Required for Maintenance of Phospho-Chk1 and G2/M Arrest during DNA Cross-Link Repair in DT40 Cells
Abstract
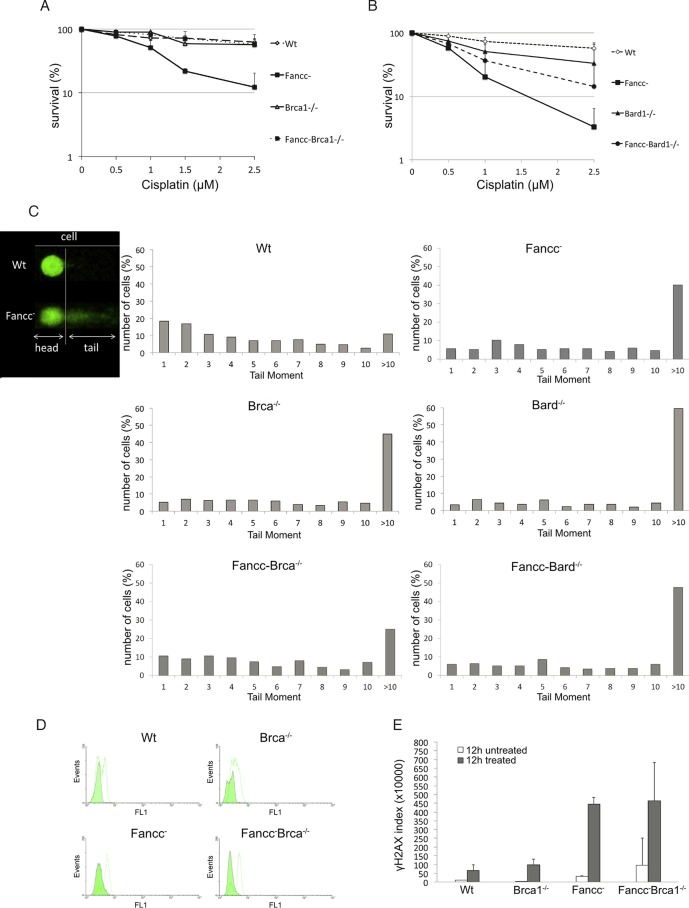
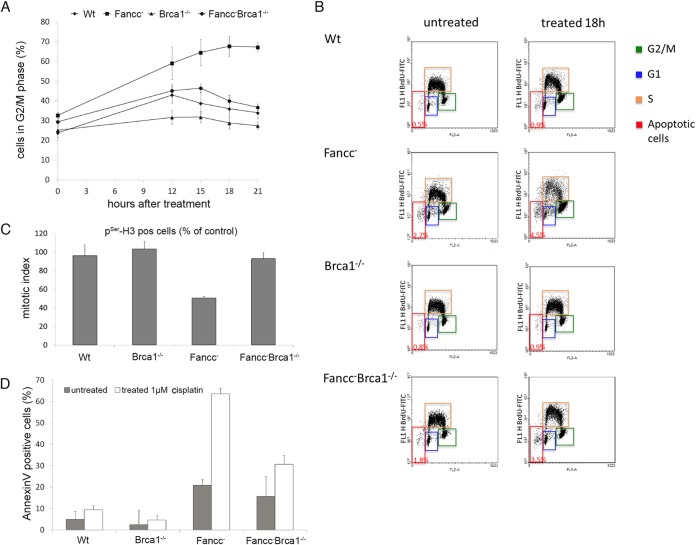
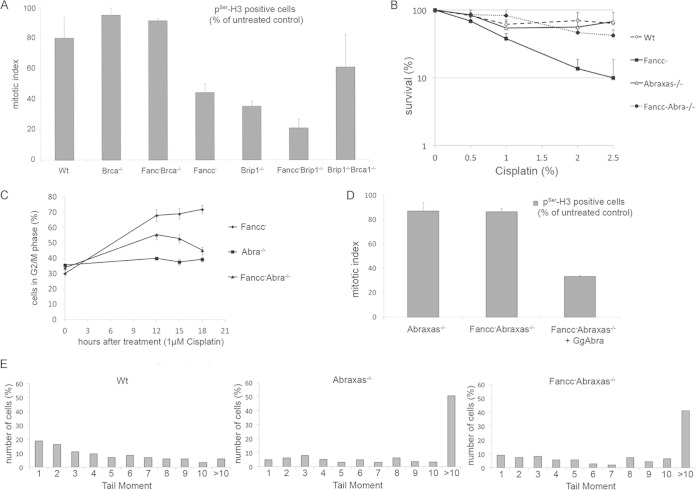
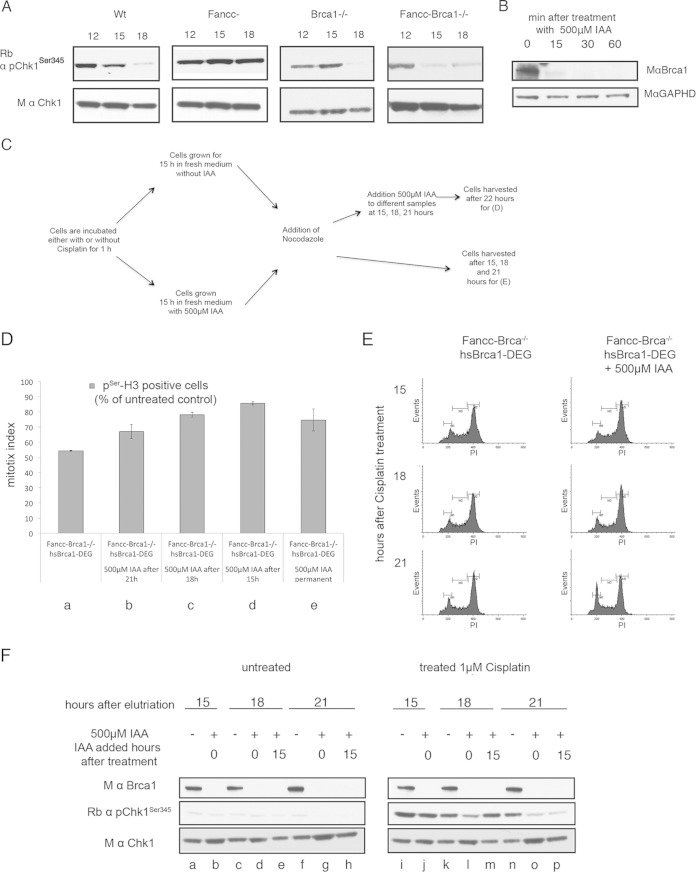
The Fanconi anemia DNA repair pathway is pivotal for the efficient repair of DNA interstrand cross-links. Here, we show that FA-defective (Fancc(-)) DT40 cells arrest in G2 phase following cross-link damage and trigger apoptosis. Strikingly, cell death was reduced in Fancc(-) cells by additional deletion of the BRCA1 tumor suppressor, resulting in elevated clonogenic survival. Increased resistance to cross-link damage was not due to loss of toxic BRCA1-mediated homologous recombination but rather through the loss of a G2 checkpoint. This proapoptotic role also required the BRCA1-A complex member ABRAXAS (FAM175A). Finally, we show that BRCA1 promotes G2 arrest and cell death by prolonging phosphorylation of Chk1 on serine 345 after DNA damage to sustain arrest. Our data imply that DNA-induced cross-link death in cells defective in the FA pathway is dependent on the ability of BRCA1 to prolong cell cycle arrest in G2 phase.
Copyright © 2015 Draga et al.
Figures

Similar articles
-
An ATR- and BRCA1-mediated Fanconi anemia pathway is required for activating the G2/M checkpoint and DNA damage repair upon rereplication.Mol Cell Biol. 2006 Jun;26(12):4601-11. doi: 10.1128/MCB.02141-05. Mol Cell Biol. 2006. PMID: 16738325 Free PMC article.
-
A role for the Fanconi anemia C protein in maintaining the DNA damage-induced G2 checkpoint.J Biol Chem. 2004 Dec 3;279(49):50986-93. doi: 10.1074/jbc.M407160200. Epub 2004 Sep 17. J Biol Chem. 2004. PMID: 15377654
-
The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair.Nat Genet. 2005 Sep;37(9):953-7. doi: 10.1038/ng1627. Epub 2005 Aug 21. Nat Genet. 2005. PMID: 16116421
-
Human syndromes with genomic instability and multiprotein machines that repair DNA double-strand breaks.Histol Histopathol. 2003 Jan;18(1):225-43. doi: 10.14670/HH-18.225. Histol Histopathol. 2003. PMID: 12507302 Review.
-
Molecular pathogenesis of fanconi anemia.Int J Hematol. 2002 Feb;75(2):123-8. doi: 10.1007/BF02982016. Int J Hematol. 2002. PMID: 11939257 Review.
Cited by
-
DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells.Nat Commun. 2017 Nov 24;8(1):1751. doi: 10.1038/s41467-017-01883-9. Nat Commun. 2017. PMID: 29170499 Free PMC article.
-
Both BRCA1-wild type and -mutant triple-negative breast cancers show sensitivity to the NAE inhibitor MLN4924 which is enhanced upon MLN4924 and cisplatin combination treatment.Oncotarget. 2020 Feb 25;11(8):784-800. doi: 10.18632/oncotarget.27485. eCollection 2020 Feb 25. Oncotarget. 2020. PMID: 32166000 Free PMC article.
-
Targeted inhibition of Polo-like kinase 1 by a novel small-molecule inhibitor induces mitotic catastrophe and apoptosis in human bladder cancer cells.J Cell Mol Med. 2017 Apr;21(4):758-767. doi: 10.1111/jcmm.13018. Epub 2016 Nov 23. J Cell Mol Med. 2017. PMID: 27878946 Free PMC article.
-
Direct regulation of Chk1 protein stability by E3 ubiquitin ligase HUWE1.FEBS J. 2020 May;287(10):1985-1999. doi: 10.1111/febs.15132. Epub 2019 Nov 29. FEBS J. 2020. PMID: 31713291 Free PMC article.
-
Integrated genomic analysis identifies novel low-frequency cis-regulatory variant rs2279658 associated with VSD risk in Chinese children.Front Cell Dev Biol. 2022 Dec 8;10:1062403. doi: 10.3389/fcell.2022.1062403. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36568976 Free PMC article.
References
-
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER III, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D'Andrea AD, Elledge SJ. 2007. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129:289–301. doi:10.1016/j.cell.2007.03.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous