

Structural insights into µ-opioid receptor activation
- PMID: 26245379
- PMCID: PMC4639397
- DOI: 10.1038/nature14886
Structural insights into µ-opioid receptor activation
Erratum in
-
Author Correction: Structural insights into μ-opioid receptor activation.Nature. 2020 Aug;584(7820):E16. doi: 10.1038/s41586-020-2542-z. Nature. 2020. PMID: 32724208
Abstract
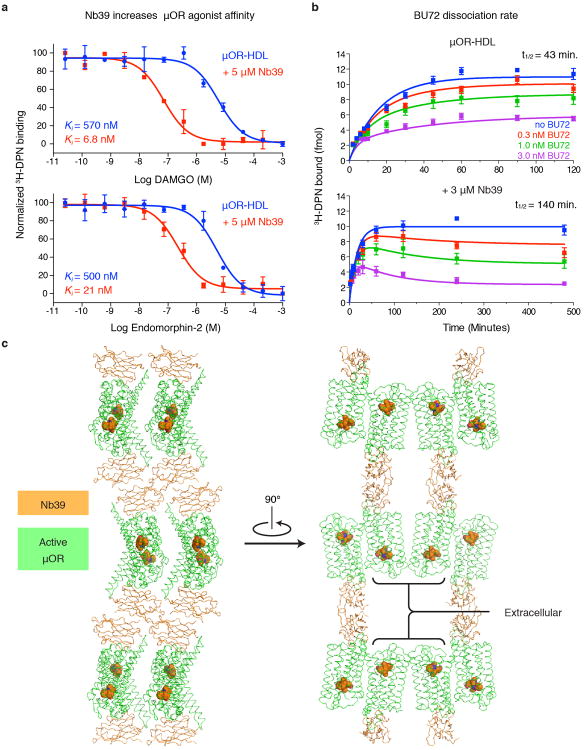
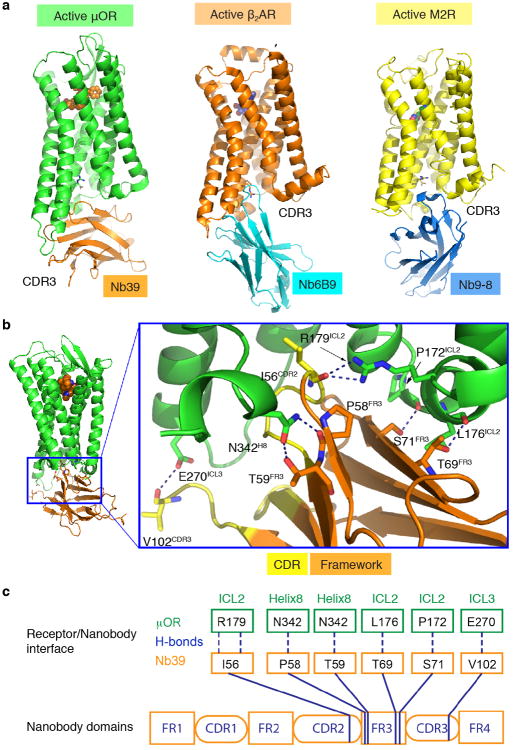
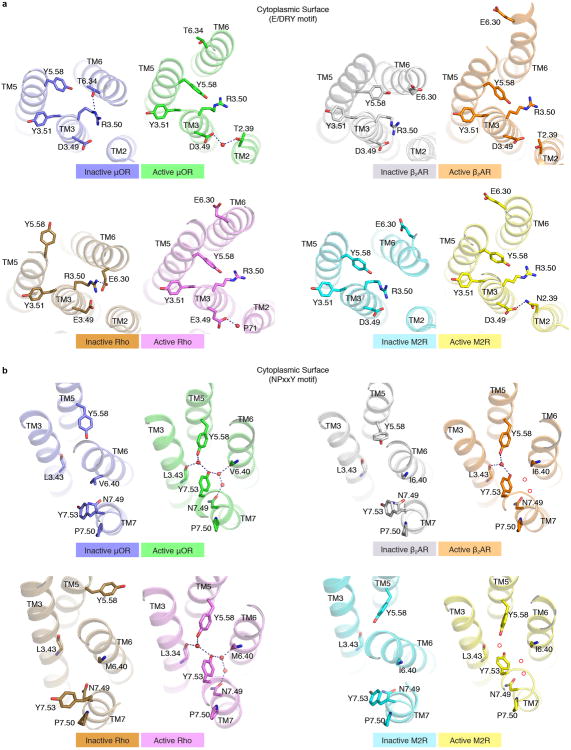
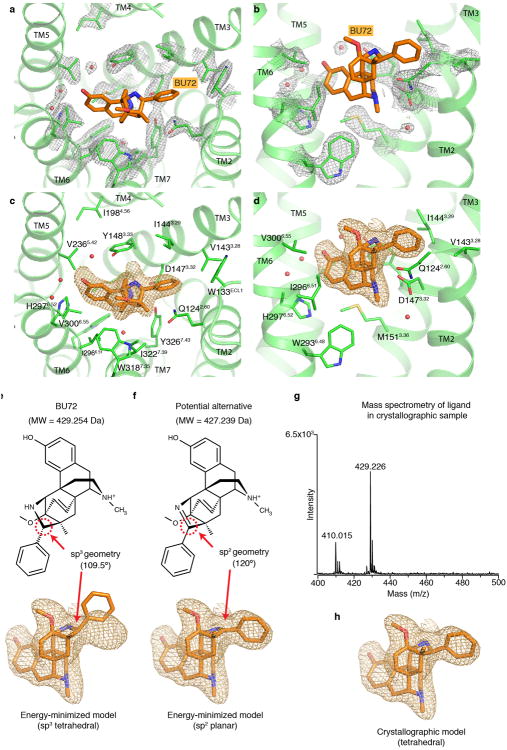
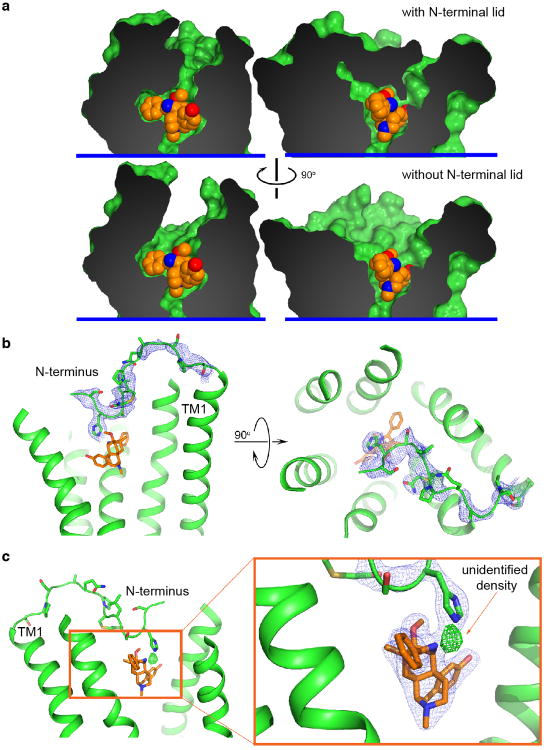
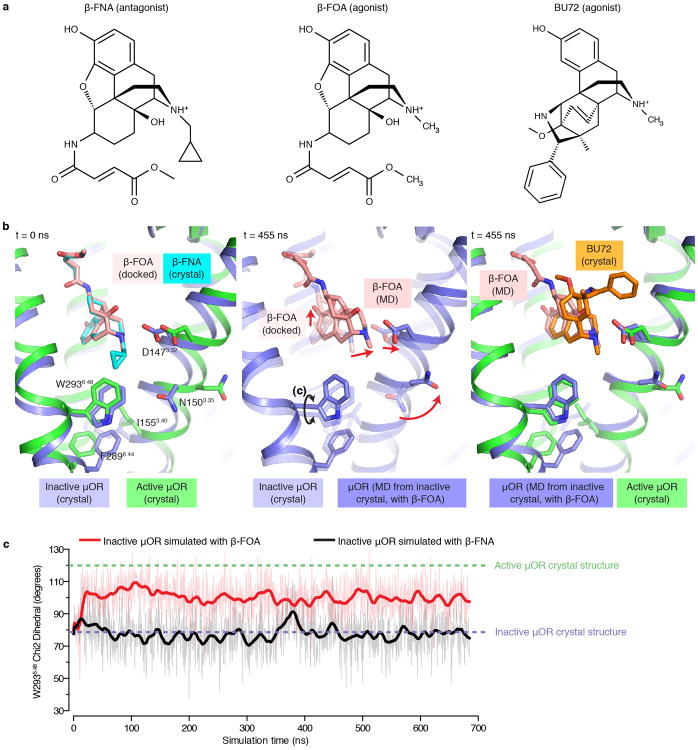
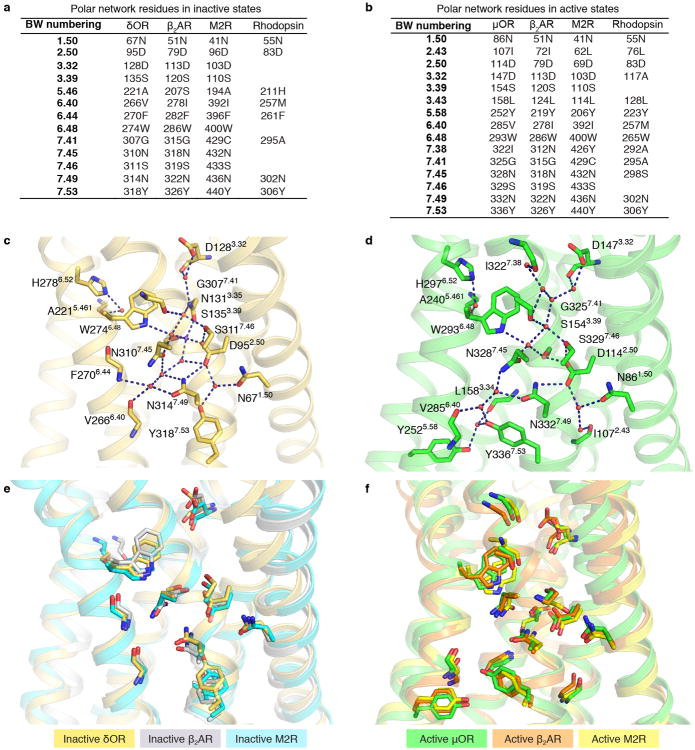
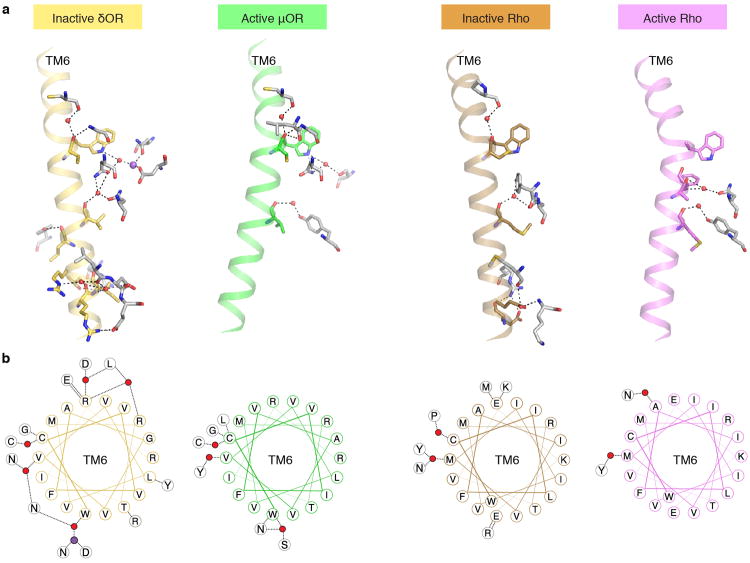
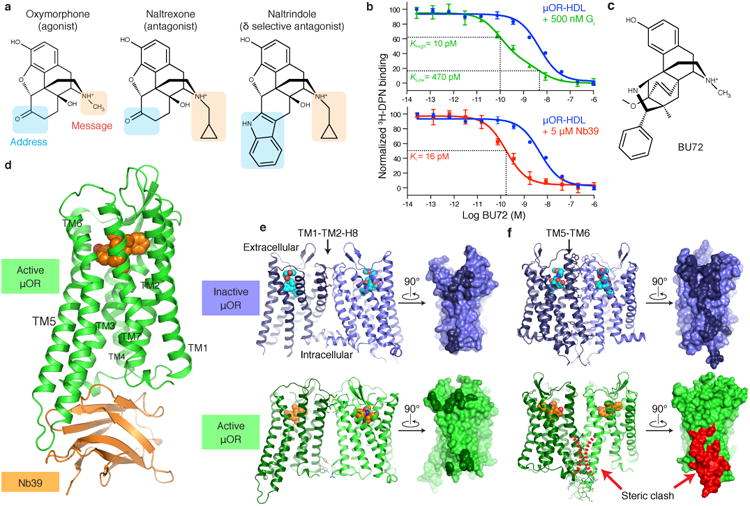
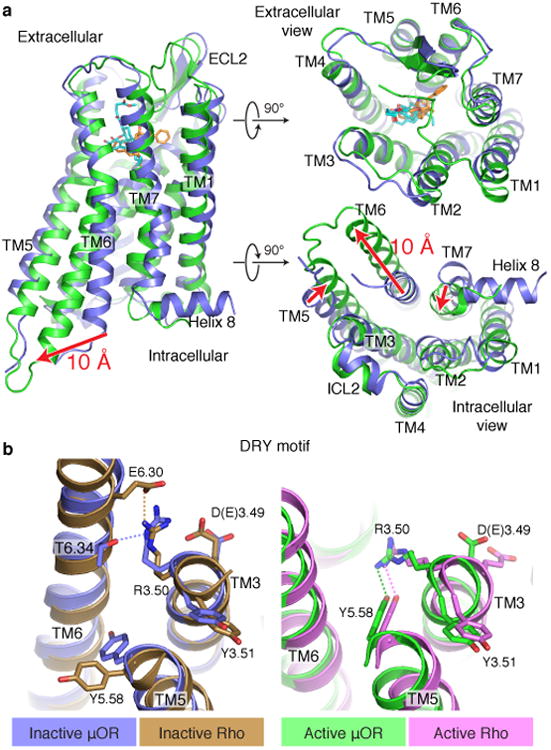
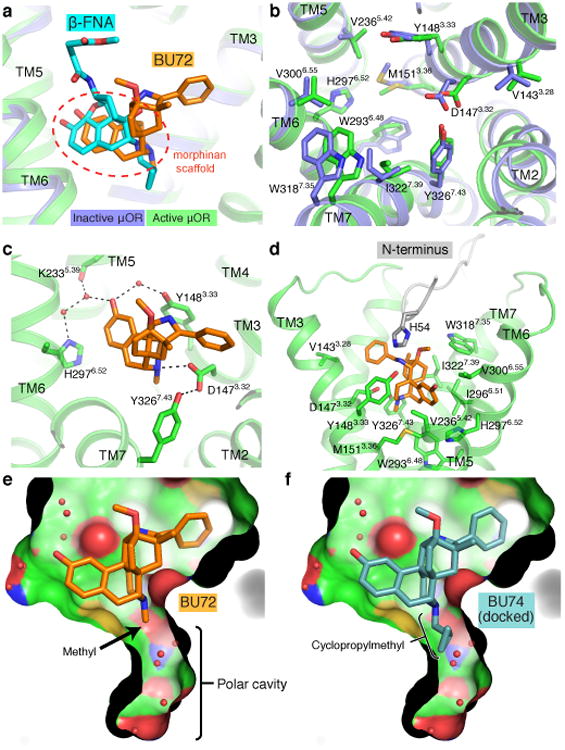
Activation of the μ-opioid receptor (μOR) is responsible for the efficacy of the most effective analgesics. To shed light on the structural basis for μOR activation, here we report a 2.1 Å X-ray crystal structure of the murine μOR bound to the morphinan agonist BU72 and a G protein mimetic camelid antibody fragment. The BU72-stabilized changes in the μOR binding pocket are subtle and differ from those observed for agonist-bound structures of the β2-adrenergic receptor (β2AR) and the M2 muscarinic receptor. Comparison with active β2AR reveals a common rearrangement in the packing of three conserved amino acids in the core of the μOR, and molecular dynamics simulations illustrate how the ligand-binding pocket is conformationally linked to this conserved triad. Additionally, an extensive polar network between the ligand-binding pocket and the cytoplasmic domains appears to play a similar role in signal propagation for all three G-protein-coupled receptors.
Figures



Similar articles
-
Propagation of conformational changes during μ-opioid receptor activation.Nature. 2015 Aug 20;524(7565):375-8. doi: 10.1038/nature14680. Epub 2015 Aug 5. Nature. 2015. PMID: 26245377 Free PMC article.
-
Structure of the µ-opioid receptor-Gi protein complex.Nature. 2018 Jun;558(7711):547-552. doi: 10.1038/s41586-018-0219-7. Epub 2018 Jun 13. Nature. 2018. PMID: 29899455 Free PMC article.
-
Computational insights into the G-protein-biased activation and inactivation mechanisms of the μ opioid receptor.Acta Pharmacol Sin. 2018 Jan;39(1):154-164. doi: 10.1038/aps.2017.158. Epub 2017 Nov 30. Acta Pharmacol Sin. 2018. PMID: 29188799 Free PMC article.
-
Structural features of β2 adrenergic receptor: crystal structures and beyond.Mol Cells. 2015;38(2):105-11. doi: 10.14348/molcells.2015.2301. Epub 2014 Dec 24. Mol Cells. 2015. PMID: 25537861 Free PMC article. Review.
-
Revealing Atomic-Level Mechanisms of Protein Allostery with Molecular Dynamics Simulations.PLoS Comput Biol. 2016 Jun 10;12(6):e1004746. doi: 10.1371/journal.pcbi.1004746. eCollection 2016 Jun. PLoS Comput Biol. 2016. PMID: 27285999 Free PMC article. Review.
Cited by
-
Investigating G-protein coupled receptor signalling with light-emitting biosensors.Front Physiol. 2024 Jan 8;14:1310197. doi: 10.3389/fphys.2023.1310197. eCollection 2023. Front Physiol. 2024. PMID: 38260094 Free PMC article. Review.
-
A combination of machine learning and infrequent metadynamics to efficiently predict kinetic rates, transition states, and molecular determinants of drug dissociation from G protein-coupled receptors.J Chem Phys. 2020 Sep 28;153(12):124105. doi: 10.1063/5.0019100. J Chem Phys. 2020. PMID: 33003748 Free PMC article.
-
The opioid receptor: emergence through millennia of pharmaceutical sciences.Front Pain Res (Lausanne). 2023 Nov 1;4:960389. doi: 10.3389/fpain.2023.960389. eCollection 2023. Front Pain Res (Lausanne). 2023. PMID: 38028425 Free PMC article. Review.
-
Smoothened transduces Hedgehog signals via activity-dependent sequestration of PKA catalytic subunits.PLoS Biol. 2021 Apr 22;19(4):e3001191. doi: 10.1371/journal.pbio.3001191. eCollection 2021 Apr. PLoS Biol. 2021. PMID: 33886552 Free PMC article.
-
Novel Dual-Target μ-Opioid Receptor and Dopamine D3 Receptor Ligands as Potential Nonaddictive Pharmacotherapeutics for Pain Management.J Med Chem. 2021 Jun 10;64(11):7778-7808. doi: 10.1021/acs.jmedchem.1c00611. Epub 2021 May 20. J Med Chem. 2021. PMID: 34011153 Free PMC article.
References
-
- Matthes HW, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. 1996 - PubMed
-
- Schumacher MA, Basbaum AI, Naidu RK. In: Basic & Clinical Pharmacology, 13e. Katzung Bertram G, Trevor Anthony J., editors. McGraw-Hill Medical; 2015.
-
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. Journal of Pharmacology and Experimental Therapeutics. 2005;314:1195–1201. - PubMed
-
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. [mu]-Opioid receptor desensitization by [beta]-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
