

TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies
- PMID: 26189817
- PMCID: PMC4573257
- DOI: 10.1016/j.ajhg.2015.06.011
TRMT5 Mutations Cause a Defect in Post-transcriptional Modification of Mitochondrial tRNA Associated with Multiple Respiratory-Chain Deficiencies
Abstract
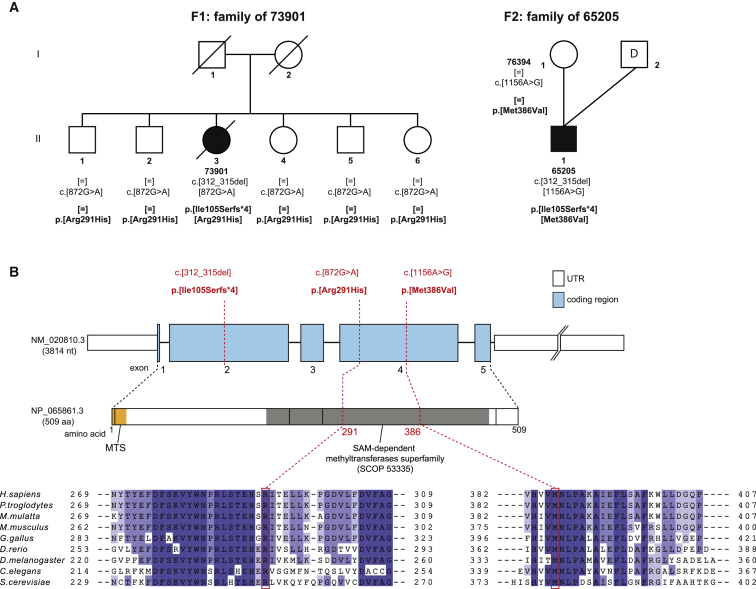
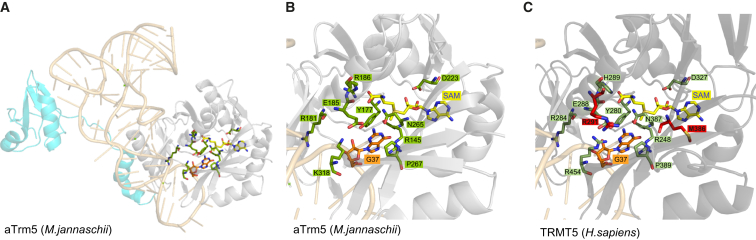
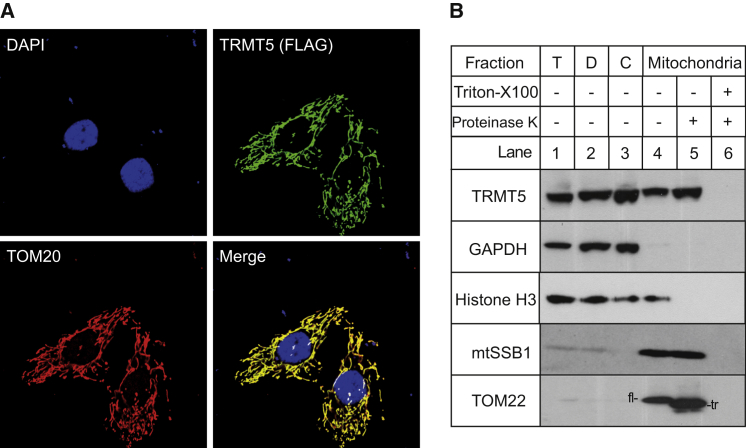
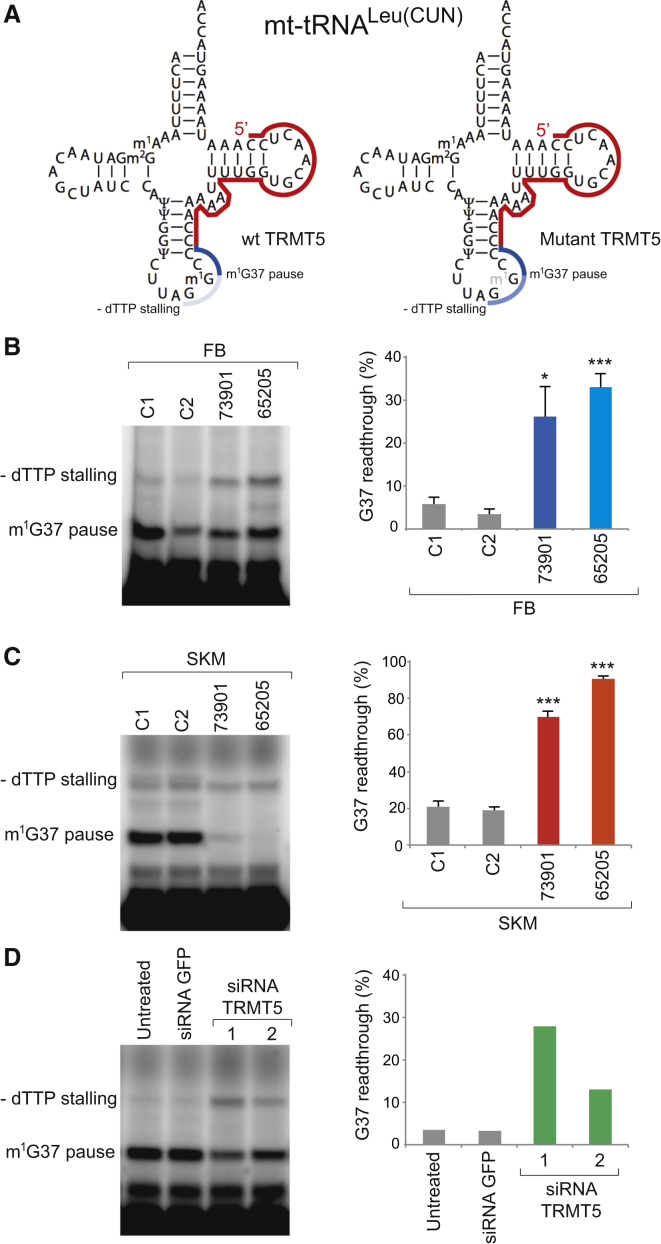
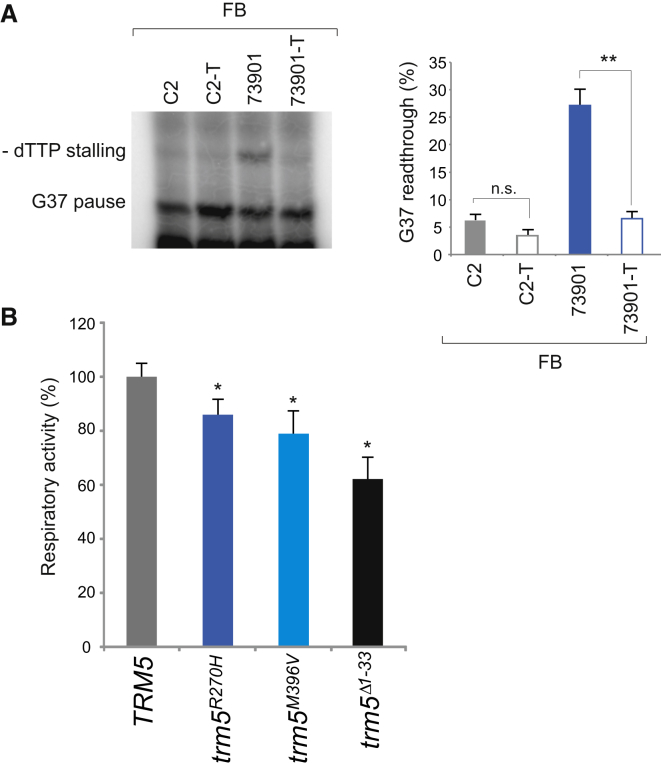
Deficiencies in respiratory-chain complexes lead to a variety of clinical phenotypes resulting from inadequate energy production by the mitochondrial oxidative phosphorylation system. Defective expression of mtDNA-encoded genes, caused by mutations in either the mitochondrial or nuclear genome, represents a rapidly growing group of human disorders. By whole-exome sequencing, we identified two unrelated individuals carrying compound heterozygous variants in TRMT5 (tRNA methyltransferase 5). TRMT5 encodes a mitochondrial protein with strong homology to members of the class I-like methyltransferase superfamily. Both affected individuals presented with lactic acidosis and evidence of multiple mitochondrial respiratory-chain-complex deficiencies in skeletal muscle, although the clinical presentation of the two affected subjects was remarkably different; one presented in childhood with failure to thrive and hypertrophic cardiomyopathy, and the other was an adult with a life-long history of exercise intolerance. Mutations in TRMT5 were associated with the hypomodification of a guanosine residue at position 37 (G37) of mitochondrial tRNA; this hypomodification was particularly prominent in skeletal muscle. Deficiency of the G37 modification was also detected in human cells subjected to TRMT5 RNAi. The pathogenicity of the detected variants was further confirmed in a heterologous yeast model and by the rescue of the molecular phenotype after re-expression of wild-type TRMT5 cDNA in cells derived from the affected individuals. Our study highlights the importance of post-transcriptional modification of mitochondrial tRNAs for faithful mitochondrial function.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies.Am J Hum Genet. 2016 May 5;98(5):993-1000. doi: 10.1016/j.ajhg.2016.03.010. Epub 2016 Apr 28. Am J Hum Genet. 2016. PMID: 27132592 Free PMC article.
-
A novel TRMT5 mutation causes a complex inherited neuropathy syndrome: The role of nerve pathology in defining a demyelinating neuropathy.Neuropathol Appl Neurobiol. 2022 Aug;48(5):e12817. doi: 10.1111/nan.12817. Epub 2022 Apr 10. Neuropathol Appl Neurobiol. 2022. PMID: 35342985
-
Mutations in GTPBP3 cause a mitochondrial translation defect associated with hypertrophic cardiomyopathy, lactic acidosis, and encephalopathy.Am J Hum Genet. 2014 Dec 4;95(6):708-20. doi: 10.1016/j.ajhg.2014.10.017. Epub 2014 Nov 26. Am J Hum Genet. 2014. PMID: 25434004 Free PMC article.
-
Posttranscriptional modifications in mitochondrial tRNA and its implication in mitochondrial translation and disease.J Biochem. 2020 Nov 1;168(5):435-444. doi: 10.1093/jb/mvaa098. J Biochem. 2020. PMID: 32818253 Review.
-
Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs.Wiley Interdiscip Rev RNA. 2011 May-Jun;2(3):376-86. doi: 10.1002/wrna.65. Epub 2011 Feb 25. Wiley Interdiscip Rev RNA. 2011. PMID: 21957023 Review.
Cited by
-
Methylation modifications in tRNA and associated disorders: Current research and potential therapeutic targets.Cell Prolif. 2024 Sep;57(9):e13692. doi: 10.1111/cpr.13692. Epub 2024 Jun 28. Cell Prolif. 2024. PMID: 38943267 Free PMC article. Review.
-
RNA modifications in physiology and disease: towards clinical applications.Nat Rev Genet. 2024 Feb;25(2):104-122. doi: 10.1038/s41576-023-00645-2. Epub 2023 Sep 15. Nat Rev Genet. 2024. PMID: 37714958 Review.
-
The role of m5C methyltransferases in cardiovascular diseases.Front Cardiovasc Med. 2023 Jul 5;10:1225014. doi: 10.3389/fcvm.2023.1225014. eCollection 2023. Front Cardiovasc Med. 2023. PMID: 37476573 Free PMC article. Review.
-
The genotypic and phenotypic spectrum of MTO1 deficiency.Mol Genet Metab. 2018 Jan;123(1):28-42. doi: 10.1016/j.ymgme.2017.11.003. Epub 2017 Nov 15. Mol Genet Metab. 2018. PMID: 29331171 Free PMC article.
-
The Pseudouridine Synthase RPUSD4 Is an Essential Component of Mitochondrial RNA Granules.J Biol Chem. 2017 Mar 17;292(11):4519-4532. doi: 10.1074/jbc.M116.771105. Epub 2017 Jan 12. J Biol Chem. 2017. PMID: 28082677 Free PMC article.
References
-
- Nicholls T.J., Rorbach J., Minczuk M. Mitochondria: mitochondrial RNA metabolism and human disease. Int. J. Biochem. Cell Biol. 2013;45:845–849. - PubMed
-
- Deutschmann A.J., Amberger A., Zavadil C., Steinbeisser H., Mayr J.A., Feichtinger R.G., Oerum S., Yue W.W., Zschocke J. Mutation or knock-down of 17β-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum. Mol. Genet. 2014;23:3618–3628. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
