

Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice
- PMID: 26077675
- PMCID: PMC4467677
- DOI: 10.1186/s12967-015-0552-7
Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice
Abstract
Background: Non-alcoholic fatty liver disease (NAFLD) is becoming a pandemic. While multiple 'hits' have been reported to contribute to NAFLD progression to non-alcoholic steatohepatitis (NASH), fibrosis and liver cancer, understanding the natural history of the specific molecular signals leading to hepatocyte damage, inflammation and fibrosis, is hampered by the lack of suitable animal models that reproduce disease progression in humans. The purpose of this study was first, to develop a mouse model that closely mimics progressive NAFLD covering the spectrum of immune, metabolic and histopathologic abnormalities present in human disease; and second, to characterize the temporal relationship between sterile/exogenous danger signals, inflammation, inflammasome activation and NAFLD progression.
Methods: Male C57Bl/6 mice were fed a high fat diet with high cholesterol and a high sugar supplement (HF-HC-HSD) for 8, 27, and 49 weeks and the extent of steatosis, liver inflammation, fibrosis and tumor development were evaluated at each time point.
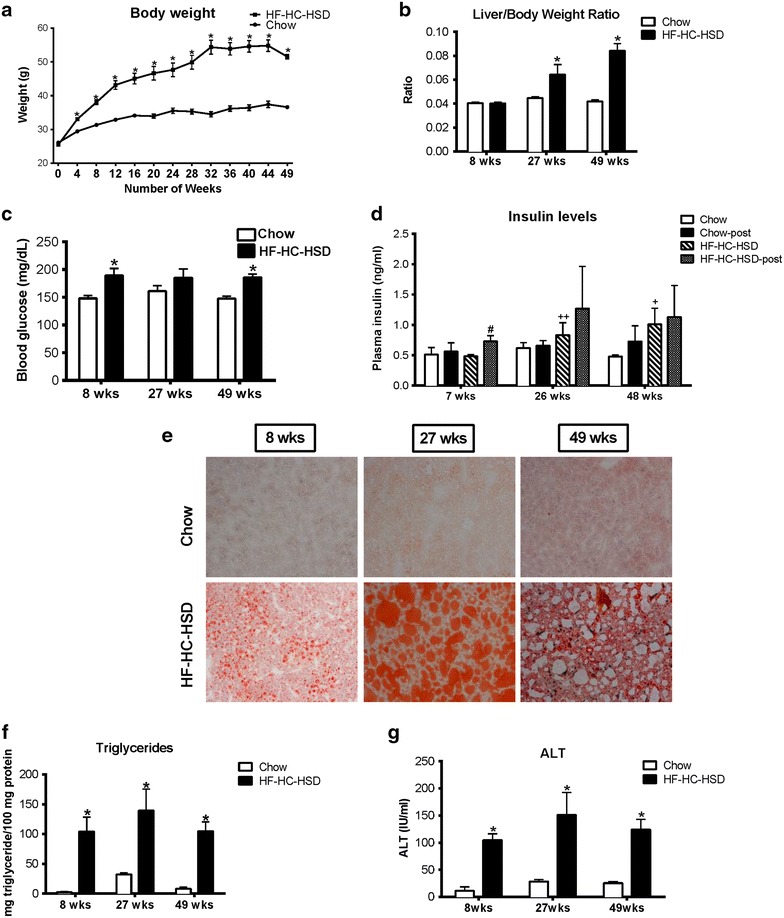
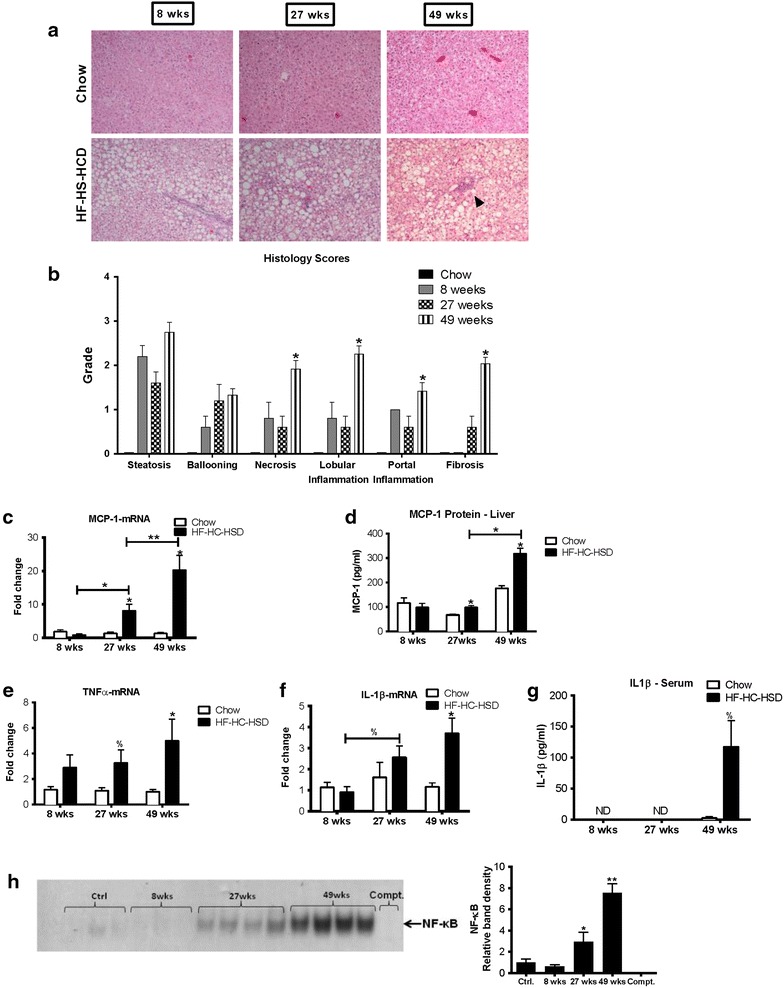
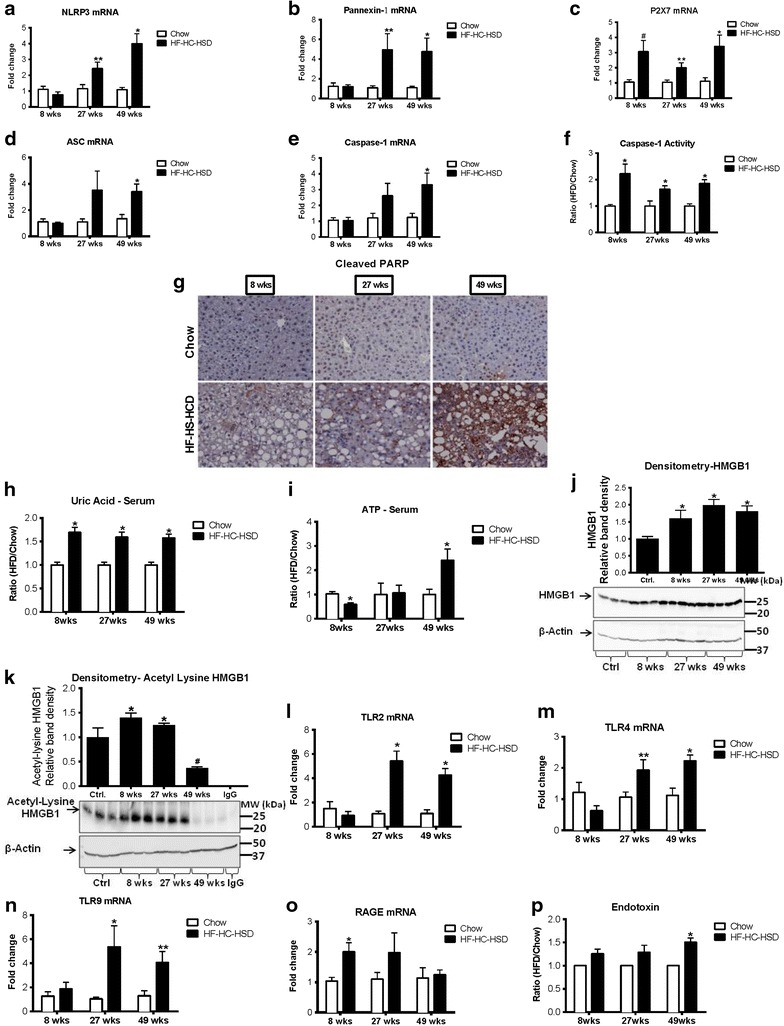
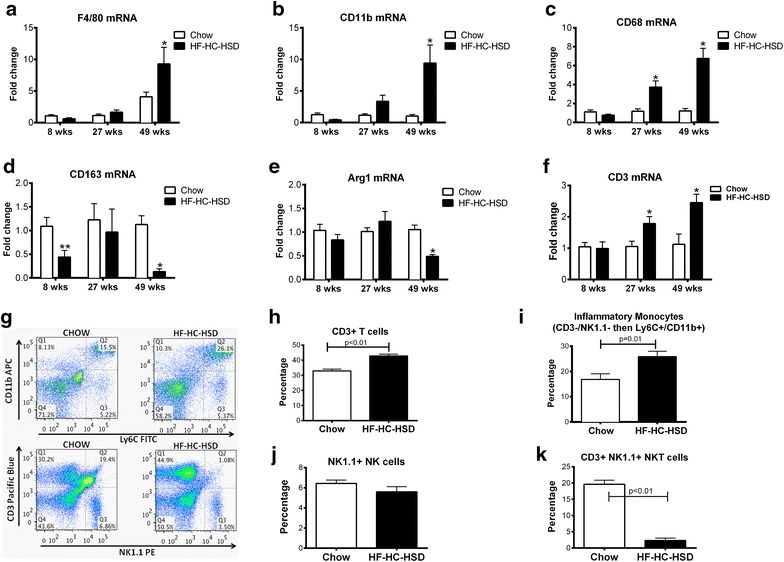
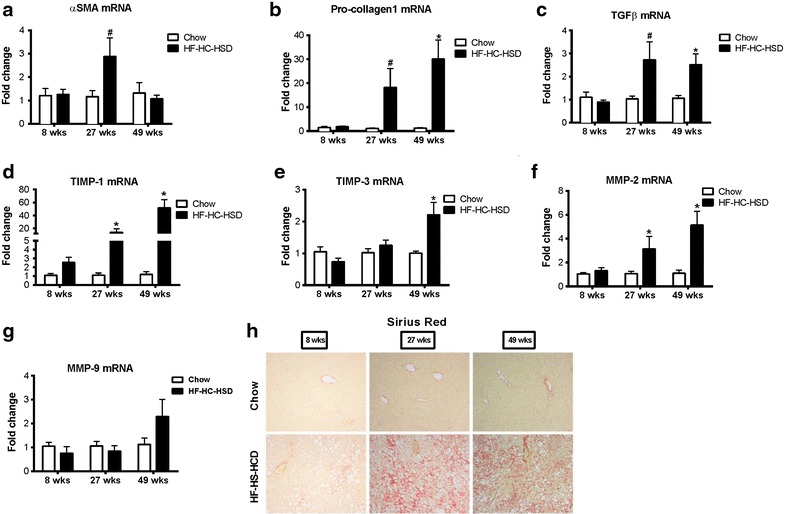
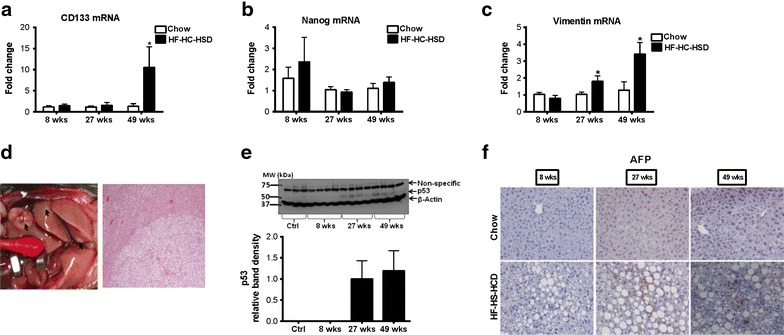
Results: The HF-HC-HSD resulted in liver steatosis at 8 weeks, progressing to steatohepatitis and early fibrosis at 27 weeks, and steatohepatitis, fibrosis, and tumor development at 49 weeks compared to chow diet. Steatohepatitis was characterized by increased levels of MCP-1, TNFα, IL-1β and increased liver NASH histological score. We found increased serum levels of sterile danger signals, uric acid and HMGB1, as early as 8 weeks, while endotoxin and ATP levels increased only after 49 weeks. Increased levels of these sterile and microbial danger signals paralleled upregulation and activation of the multiprotein complex inflammasome. At 27, 49 weeks of HF-HC-HSD, activation of M1 macrophages and loss of M2 macrophages as well as liver fibrosis were present. Finally, similar to human NASH, liver tumors occurred in 41% of mice in the absence of cirrhosis and livers expressed increased p53 and detectable AFP.
Conclusions: HF-HC-HSD over 49 weeks induces the full spectrum of liver pathophysiologic changes that characterizes the progression of NAFLD in humans. NAFLD progression to NASH, fibrosis and liver tumor follows progressive accumulation of sterile and microbial danger signals, inflammasome activation, altered M1/M2 cell ratios that likely contribute to NASH progression and hepatic tumor formation.
Figures
Similar articles
-
Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease.Obesity (Silver Spring). 2013 Jun;21(6):1189-99. doi: 10.1002/oby.20123. Epub 2013 May 10. Obesity (Silver Spring). 2013. PMID: 23666886
-
MicroRNA expression analysis in high fat diet-induced NAFLD-NASH-HCC progression: study on C57BL/6J mice.BMC Cancer. 2016 Jan 5;16:3. doi: 10.1186/s12885-015-2007-1. BMC Cancer. 2016. PMID: 26728044 Free PMC article.
-
Strain dependence of diet-induced NASH and liver fibrosis in obese mice is linked to diabetes and inflammatory phenotype.Liver Int. 2014 Aug;34(7):1084-93. doi: 10.1111/liv.12335. Epub 2013 Oct 25. Liver Int. 2014. PMID: 24107103
-
Utilization of animal models to investigate nonalcoholic steatohepatitis-associated hepatocellular carcinoma.Oncotarget. 2016 Jul 5;7(27):42762-42776. doi: 10.18632/oncotarget.8641. Oncotarget. 2016. PMID: 27072576 Free PMC article. Review.
-
Galectin-3 and IL-33/ST2 axis roles and interplay in diet-induced steatohepatitis.World J Gastroenterol. 2016 Nov 28;22(44):9706-9717. doi: 10.3748/wjg.v22.i44.9706. World J Gastroenterol. 2016. PMID: 27956794 Free PMC article. Review.
Cited by
-
Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases.Oxid Med Cell Longev. 2016;2016:4234061. doi: 10.1155/2016/4234061. Epub 2016 Dec 14. Oxid Med Cell Longev. 2016. PMID: 28070230 Free PMC article. Review.
-
Hepatocellular carcinoma is accelerated by NASH involving M2 macrophage polarization mediated by hif-1αinduced IL-10.Oncoimmunology. 2016 Sep 26;5(10):e1221557. doi: 10.1080/2162402X.2016.1221557. eCollection 2016. Oncoimmunology. 2016. PMID: 27853646 Free PMC article.
-
Role of FXR in Liver Inflammation during Nonalcoholic Steatohepatitis.Curr Pharmacol Rep. 2017 Apr;3(2):92-100. doi: 10.1007/s40495-017-0085-2. Epub 2017 Feb 21. Curr Pharmacol Rep. 2017. PMID: 28983452 Free PMC article.
-
An Overview of Murine High Fat Diet as a Model for Type 2 Diabetes Mellitus.J Diabetes Res. 2016;2016:2902351. doi: 10.1155/2016/2902351. Epub 2016 Jul 31. J Diabetes Res. 2016. PMID: 27547764 Free PMC article. Review.
-
Opisthorchis viverrini Infection Augments the Severity of Nonalcoholic Fatty Liver Disease in High-Fat/High-Fructose Diet-Fed Hamsters.Am J Trop Med Hyg. 2019 Nov;101(5):1161-1169. doi: 10.4269/ajtmh.19-0442. Am J Trop Med Hyg. 2019. PMID: 31482785 Free PMC article.
References
-
- Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. doi: 10.1016/S0140-6736(14)60460-8. - DOI - PMC - PubMed
-
- Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592–1609. doi: 10.1053/j.gastro.2012.04.001. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
