

H3K27 Demethylation at the Proviral Promoter Sensitizes Latent HIV to the Effects of Vorinostat in Ex Vivo Cultures of Resting CD4+ T Cells
- PMID: 26041287
- PMCID: PMC4524215
- DOI: 10.1128/JVI.00572-15
H3K27 Demethylation at the Proviral Promoter Sensitizes Latent HIV to the Effects of Vorinostat in Ex Vivo Cultures of Resting CD4+ T Cells
Abstract
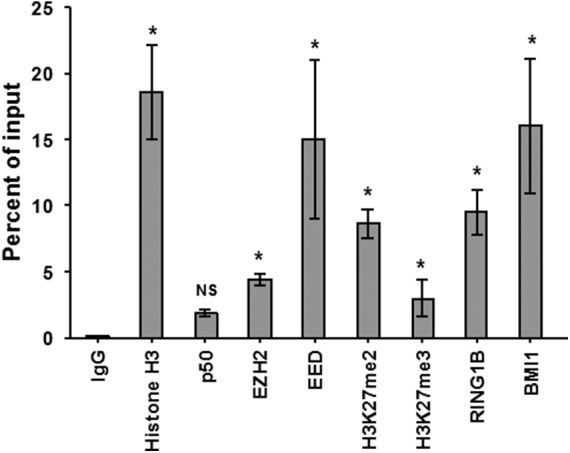
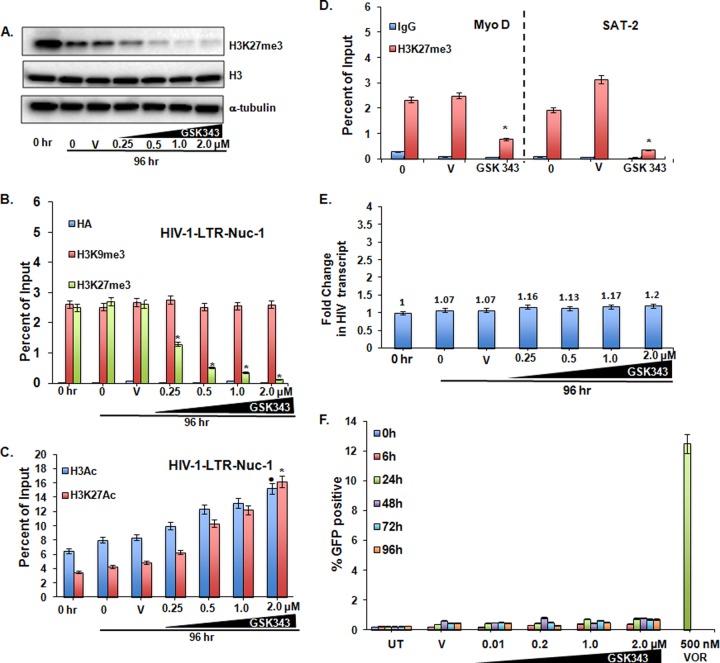
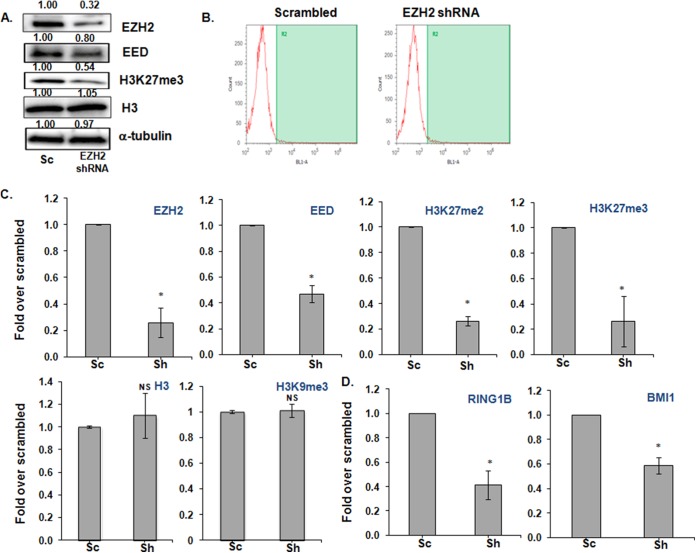
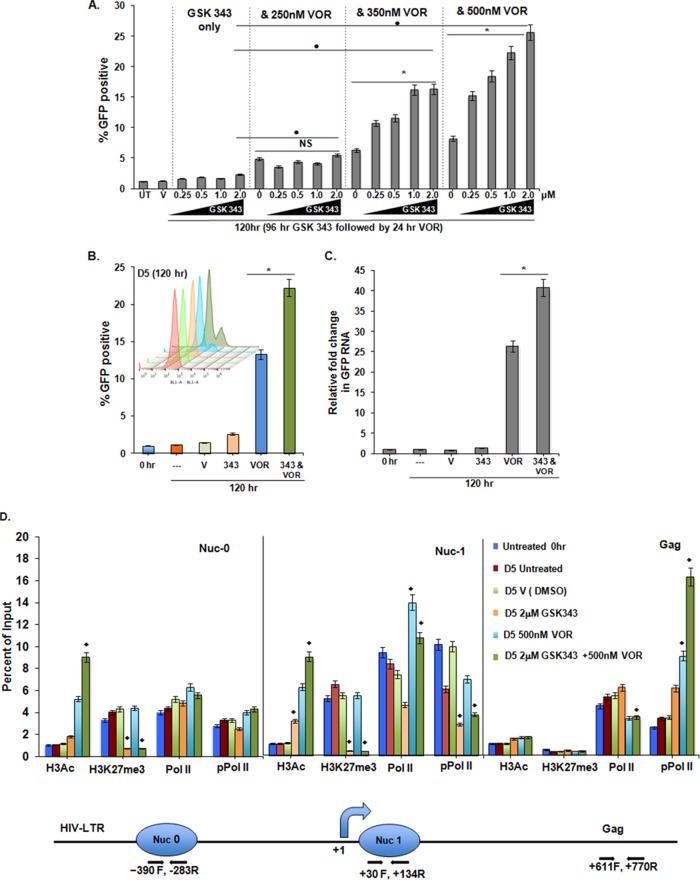
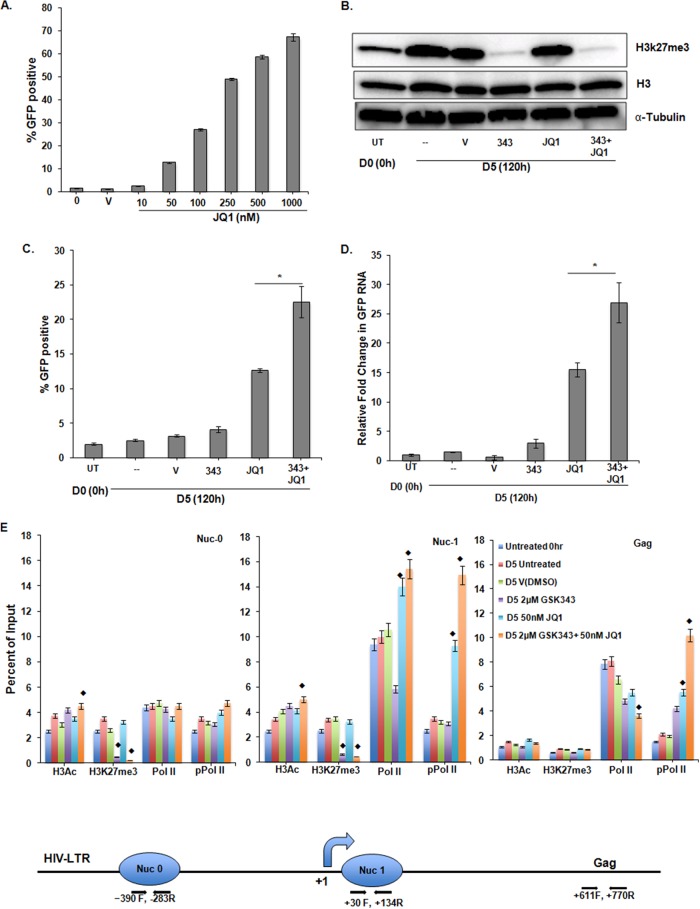
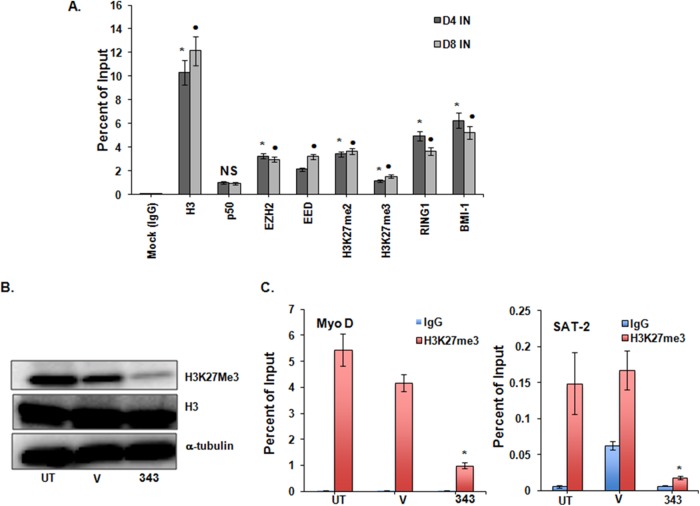
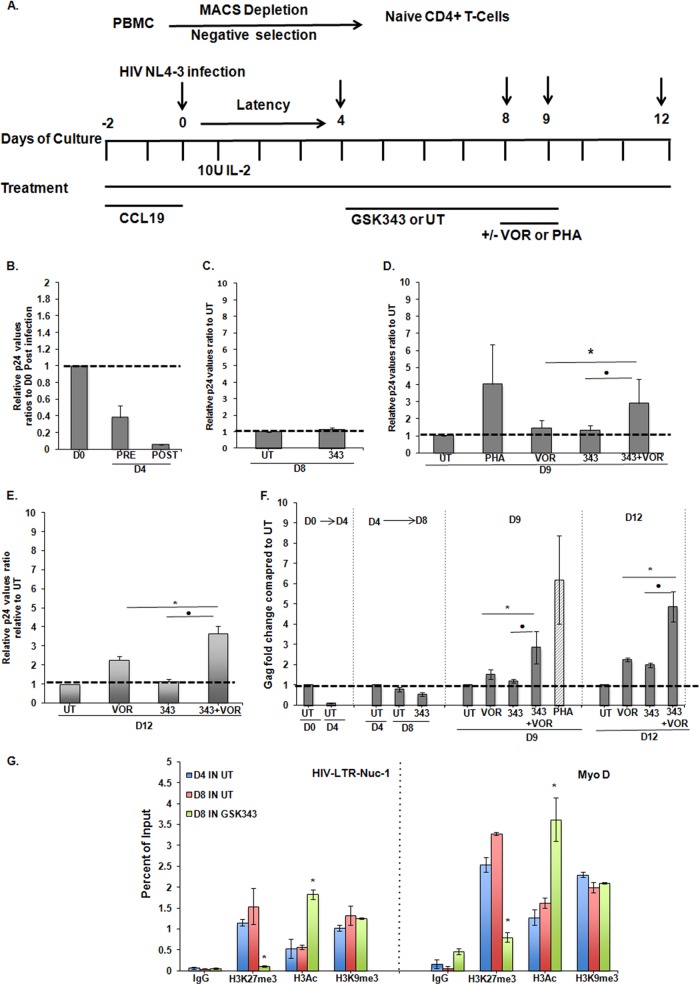
Histone methyltransferase inhibitors (HMTis) and histone deacetylase inhibitors (HDACis) are reported to synergistically induce the expression of latent human immunodeficiency virus type 1 (HIV-1), but studies have largely been performed with cell lines. As specific and potent HMTis directed at EZH1 (enhancer of zeste 2 Polycomb repressive complex 2 subunit 1)/EZH2 are now in human testing, we wished to rigorously test such an inhibitor in a primary resting T-cell model of HIV latency. We found that GSK343, a potent and selective EZH2/EZH1 inhibitor, reduced trimethylation of histone 3 at lysine 27 (H3K27) of the HIV provirus in resting cells. Remarkably, this epigenetic change was not associated with increased proviral expression in latently infected resting cells. However, following the reduction in H3K27 at the HIV long terminal repeat (LTR), subsequent exposure to the HDACi suberoylanilide hydroxamic acid or vorinostat (VOR) resulted in increases in HIV gag RNA and HIV p24 antigen production that were up to 2.5-fold greater than those induced by VOR alone. Therefore, in primary resting CD4(+) T cells, true mechanistic synergy in the reversal of HIV latency may be achieved by the combination of HMTis and HDACis. Although other cellular effects of EZH2 inhibition may contribute to the sensitization of the HIV LTR to subsequent exposure to VOR, and to increase viral antigen production, this synergistic effect is directly associated with H3K27 demethylation at nucleosome 1 (Nuc-1). Based upon our findings, the combination of HMTis and HDACis should be considered for testing in animal models or clinical trials.
Importance: Demethylation of H3K27 mediated by the histone methyltransferase inhibitor GSK343 in primary resting T cells is slow, occurring over 96 h, but by itself does not result in a significant upregulation of cell-associated HIV RNA expression or viral antigen production. However, following H3K27 demethylation, latent viral expression within infected primary resting CD4(+) T cells is synergistically increased upon exposure to the histone deacetylase inhibitor vorinostat. Demethylation at H3K27 sensitizes the HIV promoter to the effects of an HDACi and provides a proof-of-concept for the testing of combination epigenetic approaches to disrupt latent HIV infection, a necessary step toward the eradication of HIV infection.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency.mBio. 2017 Feb 28;8(1):e00133-17. doi: 10.1128/mBio.00133-17. mBio. 2017. PMID: 28246360 Free PMC article.
-
Cellular Gene Modulation of HIV-Infected CD4 T Cells in Response to Serial Treatment with the Histone Deacetylase Inhibitor Vorinostat.J Virol. 2020 Jun 16;94(13):e00351-20. doi: 10.1128/JVI.00351-20. Print 2020 Jun 16. J Virol. 2020. PMID: 32295913 Free PMC article.
-
Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2.J Virol. 2011 Sep;85(17):9078-89. doi: 10.1128/JVI.00836-11. Epub 2011 Jun 29. J Virol. 2011. PMID: 21715480 Free PMC article.
-
HIV-Induced Epigenetic Alterations in Host Cells.Adv Exp Med Biol. 2016;879:27-38. doi: 10.1007/978-3-319-24738-0_2. Adv Exp Med Biol. 2016. PMID: 26659262 Review.
-
JMJD3 as an epigenetic regulator in development and disease.Int J Biochem Cell Biol. 2015 Oct;67:148-57. doi: 10.1016/j.biocel.2015.07.006. Epub 2015 Jul 17. Int J Biochem Cell Biol. 2015. PMID: 26193001 Free PMC article. Review.
Cited by
-
Regulation of the expression of proinflammatory cytokines induced by SARS-CoV-2.World J Clin Cases. 2021 Mar 6;9(7):1513-1523. doi: 10.12998/wjcc.v9.i7.1513. World J Clin Cases. 2021. PMID: 33728295 Free PMC article. Review.
-
Envelope-specific antibodies and antibody-derived molecules for treating and curing HIV infection.Nat Rev Drug Discov. 2016 Dec;15(12):823-834. doi: 10.1038/nrd.2016.173. Epub 2016 Oct 7. Nat Rev Drug Discov. 2016. PMID: 27725635 Free PMC article. Review.
-
HTLV-1: Regulating the Balance Between Proviral Latency and Reactivation.Front Microbiol. 2018 Mar 19;9:449. doi: 10.3389/fmicb.2018.00449. eCollection 2018. Front Microbiol. 2018. PMID: 29615991 Free PMC article. Review.
-
HIV Provirus Stably Reproduces Parental Latent and Induced Transcription Phenotypes Regardless of the Chromosomal Integration Site.J Virol. 2016 May 12;90(11):5302-14. doi: 10.1128/JVI.02842-15. Print 2016 Jun 1. J Virol. 2016. PMID: 26984732 Free PMC article.
-
Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency.mBio. 2017 Feb 28;8(1):e00133-17. doi: 10.1128/mBio.00133-17. mBio. 2017. PMID: 28246360 Free PMC article.
References
-
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi:10.1126/science.278.5341.1295. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
