

Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2α signaling pathways and implications for autophagy
- PMID: 26039820
- PMCID: PMC4614032
- DOI: 10.1080/15384101.2015.1056947
Cellular adaptation to nutrient deprivation: crosstalk between the mTORC1 and eIF2α signaling pathways and implications for autophagy
Abstract
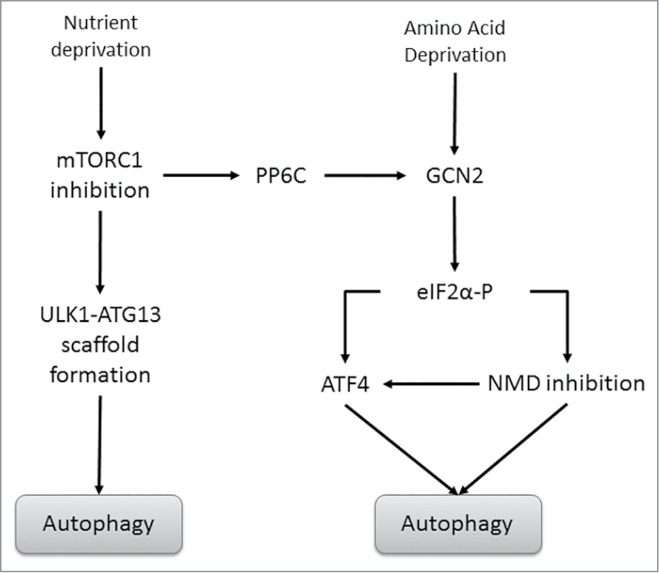
The hostile tumor microenvironment results in the generation of intracellular stresses including hypoxia and nutrient deprivation. In order to adapt to such conditions, the cell utilizes several stress-response mechanisms, including the attenuation of protein synthesis, the inhibition of cellular proliferation, and induction of autophagy. Autophagy leads to the degradation of cellular contents, including damaged organelles and mutant proteins, which the cell can then use as an alternate energy source. Two integral changes to the signaling milieu to promote such a response include inhibition of the mammalian target of rapamycin complex 1 (mTORC1) and phosphorylation of eIF2α. This review will describe how conditions found in the tumor microenvironment regulate mTORC1 as well as eIF2α, the downstream impact of these modifications, and the implications in tumorigenesis. We will then discuss the remarkable similarities and overlapping function of these 2 signaling pathways, focusing on the response to amino acid deprivation, and present a new model involving crosstalk between them based on our recent work.
Keywords: PP6C; autophagy; eIF2α; integrated stress response; mTORC1; unfolded protein response.
Figures


Similar articles
-
Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma.Sci Signal. 2015 Mar 10;8(367):ra27. doi: 10.1126/scisignal.aaa0899. Sci Signal. 2015. PMID: 25759478 Free PMC article.
-
Autophagy modulates amino acid signaling network in myotubes: differential effects on mTORC1 pathway and the integrated stress response.FASEB J. 2015 Feb;29(2):394-407. doi: 10.1096/fj.14-252841. Epub 2014 Nov 5. FASEB J. 2015. PMID: 25376834
-
Nutrient-sensing mTORC1: Integration of metabolic and autophagic signals.J Mol Cell Cardiol. 2016 Jun;95:31-41. doi: 10.1016/j.yjmcc.2016.01.005. Epub 2016 Jan 7. J Mol Cell Cardiol. 2016. PMID: 26773603 Free PMC article. Review.
-
ATM engages the TSC2/mTORC1 signaling node to regulate autophagy.Autophagy. 2010 Jul;6(5):672-3. doi: 10.4161/auto.6.5.12509. Epub 2010 Jul 1. Autophagy. 2010. PMID: 20581436 Free PMC article.
-
Autophagy regulation by nutrient signaling.Cell Res. 2014 Jan;24(1):42-57. doi: 10.1038/cr.2013.166. Epub 2013 Dec 17. Cell Res. 2014. PMID: 24343578 Free PMC article. Review.
Cited by
-
Lysosomes, Autophagy, and Hormesis in Cell Physiology, Pathology, and Age-Related Disease.Dose Response. 2020 Jul 7;18(3):1559325820934227. doi: 10.1177/1559325820934227. eCollection 2020 Jul-Sep. Dose Response. 2020. PMID: 32684871 Free PMC article. Review.
-
Autophagy inhibition enhances RAD001-induced cytotoxicity in human bladder cancer cells.Drug Des Devel Ther. 2016 Apr 18;10:1501-13. doi: 10.2147/DDDT.S95900. eCollection 2016. Drug Des Devel Ther. 2016. PMID: 27143856 Free PMC article.
-
Mammalian integrated stress responses in stressed organelles and their functions.Acta Pharmacol Sin. 2024 Jun;45(6):1095-1114. doi: 10.1038/s41401-023-01225-0. Epub 2024 Jan 24. Acta Pharmacol Sin. 2024. PMID: 38267546 Review.
-
Unbiased translation proteomics upon cell stress.Mol Cell Oncol. 2020 May 18;7(4):1763150. doi: 10.1080/23723556.2020.1763150. eCollection 2020. Mol Cell Oncol. 2020. PMID: 32944625 Free PMC article.
-
The Role of mTOR and eIF Signaling in Benign Endometrial Diseases.Int J Mol Sci. 2022 Mar 22;23(7):3416. doi: 10.3390/ijms23073416. Int J Mol Sci. 2022. PMID: 35408777 Free PMC article. Review.
References
-
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 110:177–89; PMID:12150926; http://dx.doi.org/10.1016/S0092-8674(02)00833-4 - DOI - PubMed
-
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol cell 2003; 11:895–904; PMID:12718876; http://dx.doi.org/10.1016/S1097-2765(03)00114-X - DOI - PubMed
-
- Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 2007; 9:316–23; PMID:17277771; http://dx.doi.org/10.1038/ncb1547 - DOI - PubMed
-
- Hardie DG, Carling D, Carlson M. The AMP-activated/SNF1 protein kinase subfamily: metabolic sensors of the eukaryotic cell? Ann Rev Biochem 1998; 67:821–55; PMID:9759505; http://dx.doi.org/10.1146/annurev.biochem.67.1.821 - DOI - PubMed
-
- Oakhill JS, Scott JW, Kemp BE. Structure and function of AMP-activated protein kinase. Acta Physiol 2009; 196:3–14; PMID:19245650; http://dx.doi.org/10.1111/j.1748-1716.2009.01977.x - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources