

Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP
- PMID: 26028026
- PMCID: PMC4666838
- DOI: 10.1038/onc.2015.179
Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP
Abstract
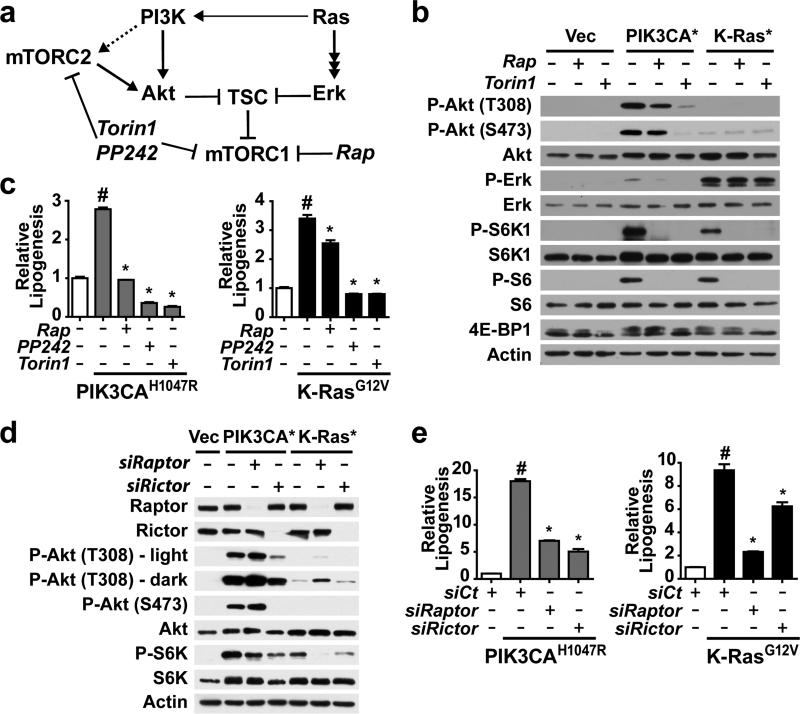
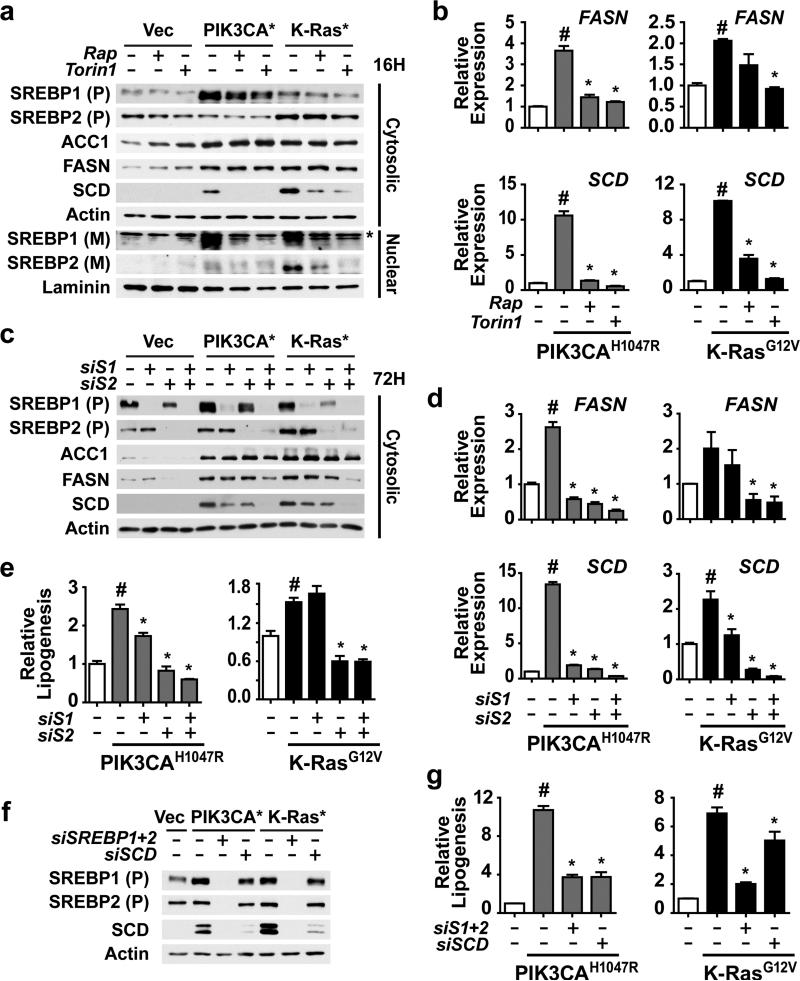
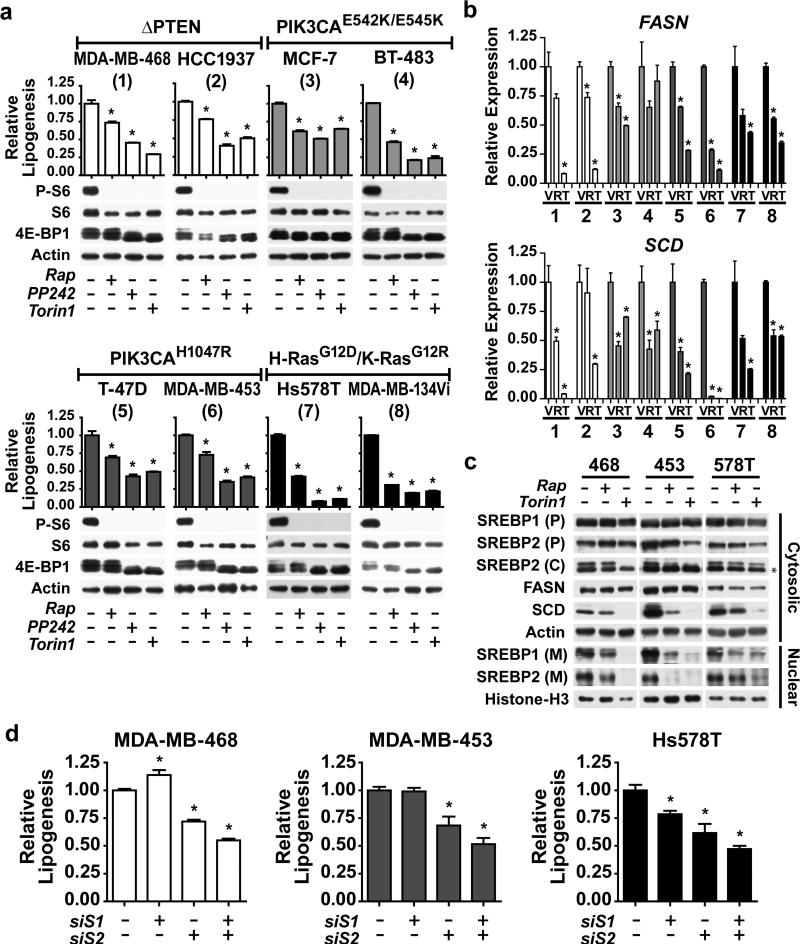
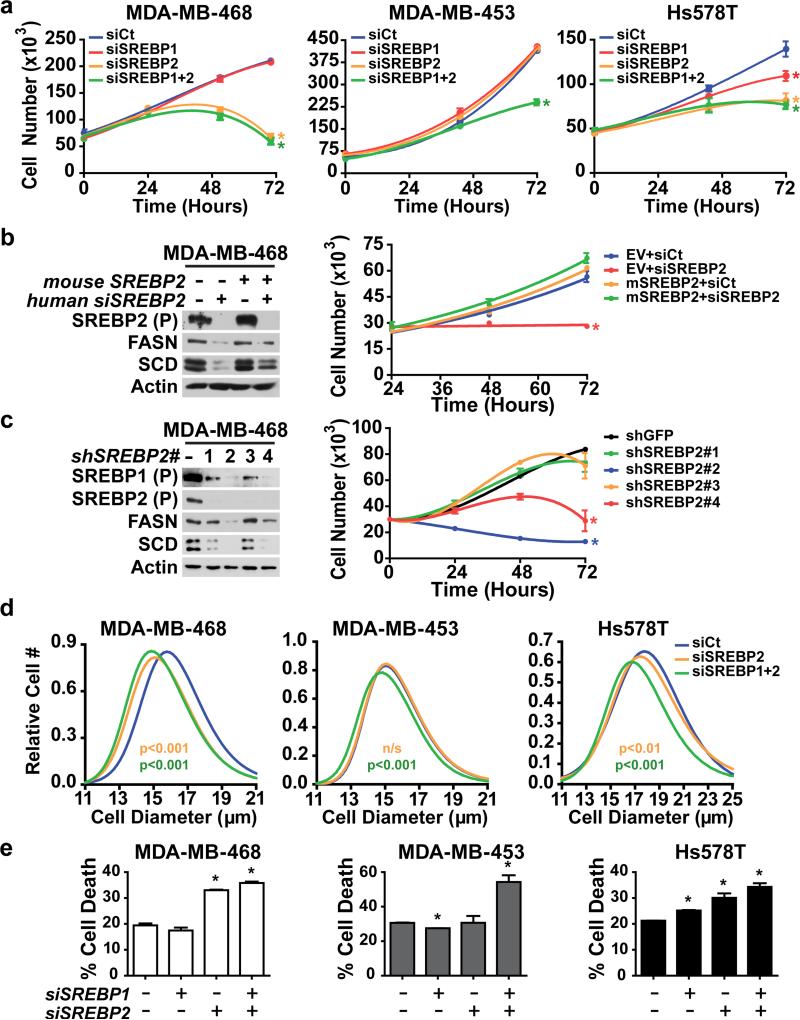
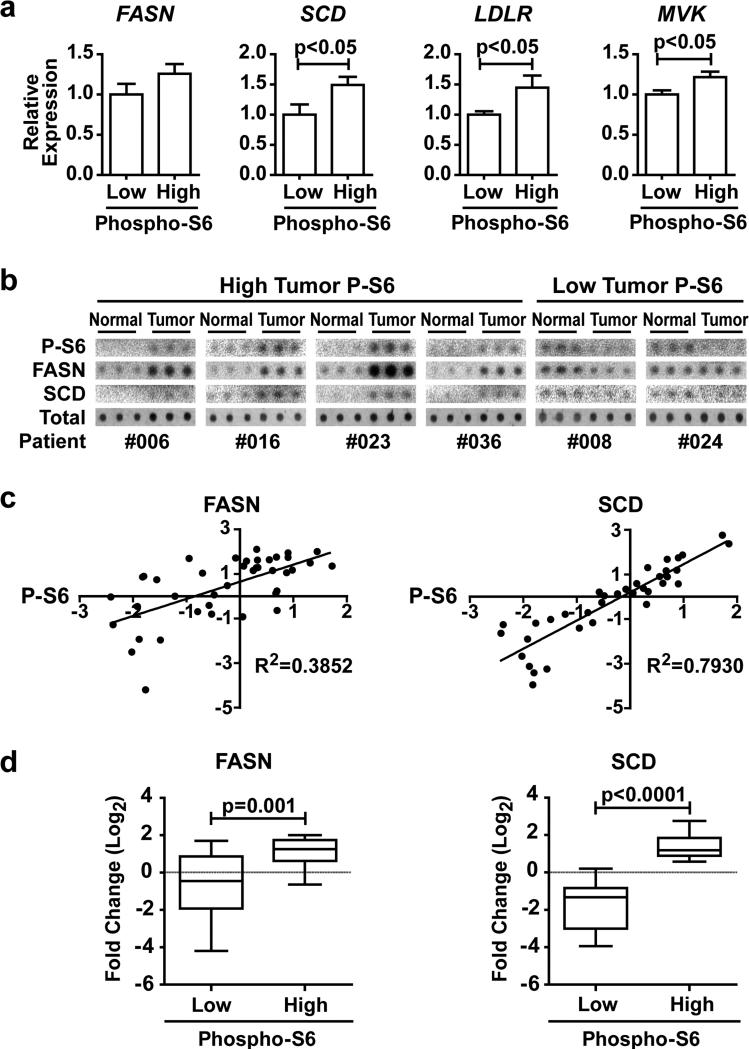
An enhanced capacity for de novo lipid synthesis is a metabolic feature of most cancer cells that distinguishes them from their cells of origin. However, the mechanisms through which oncogenes alter lipid metabolism are poorly understood. We find that expression of oncogenic PI3K (H1047R) or K-Ras (G12V) in breast epithelial cells is sufficient to induce de novo lipogenesis, and this occurs through the convergent activation of the mechanistic target of rapamycin complex 1 (mTORC1) downstream of these common oncogenes. Oncogenic stimulation of mTORC1 signaling in this isogenic setting or a panel of eight breast cancer cell lines leads to activation of the sterol regulatory element-binding proteins (SREBP1 and SREBP2) that are required for oncogene-induced lipid synthesis. The SREBPs are also required for the growth factor-independent growth and proliferation of oncogene-expressing cells. Finally, we find that elevated mTORC1 signaling is associated with increased mRNA and protein levels of canonical SREBP targets in primary human breast cancer samples. These data suggest that the mTORC1/SREBP pathway is a major mechanism through which common oncogenic signaling events induce de novo lipid synthesis to promote aberrant growth and proliferation of cancer cells.
Figures

Similar articles
-
Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma.Gastroenterology. 2011 Mar;140(3):1071-83. doi: 10.1053/j.gastro.2010.12.006. Epub 2010 Dec 11. Gastroenterology. 2011. PMID: 21147110 Free PMC article.
-
Connecting mTORC1 signaling to SREBP-1 activation.Curr Opin Lipidol. 2012 Jun;23(3):226-234. doi: 10.1097/MOL.0b013e328352dd03. Curr Opin Lipidol. 2012. PMID: 22449814 Review.
-
FGFR3 stimulates stearoyl CoA desaturase 1 activity to promote bladder tumor growth.Cancer Res. 2012 Nov 15;72(22):5843-55. doi: 10.1158/0008-5472.CAN-12-1329. Epub 2012 Sep 26. Cancer Res. 2012. PMID: 23019225
-
Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis.Proc Natl Acad Sci U S A. 2020 Dec 8;117(49):31189-31197. doi: 10.1073/pnas.2017152117. Epub 2020 Nov 23. Proc Natl Acad Sci U S A. 2020. PMID: 33229547 Free PMC article.
-
Regulation of the SREBP transcription factors by mTORC1.Biochem Soc Trans. 2011 Apr;39(2):495-9. doi: 10.1042/BST0390495. Biochem Soc Trans. 2011. PMID: 21428927 Review.
Cited by
-
Reprogramming of Lipid Metabolism in Lung Cancer: An Overview with Focus on EGFR-Mutated Non-Small Cell Lung Cancer.Cells. 2022 Jan 25;11(3):413. doi: 10.3390/cells11030413. Cells. 2022. PMID: 35159223 Free PMC article. Review.
-
Targeting lipid metabolism in the treatment of ovarian cancer.Oncotarget. 2022 May 25;13:768-783. doi: 10.18632/oncotarget.28241. eCollection 2022. Oncotarget. 2022. PMID: 35634242 Free PMC article. Review.
-
Comprehensive Transcriptomic Analysis of Novel Class I HDAC Proteolysis Targeting Chimeras (PROTACs).Biochemistry. 2023 Feb 7;62(3):645-656. doi: 10.1021/acs.biochem.2c00288. Epub 2022 Aug 10. Biochemistry. 2023. PMID: 35948047 Free PMC article.
-
Lipid metabolism reprogramming and its potential targets in cancer.Cancer Commun (Lond). 2018 May 21;38(1):27. doi: 10.1186/s40880-018-0301-4. Cancer Commun (Lond). 2018. PMID: 29784041 Free PMC article. Review.
-
In vivo CRISPR screening identifies geranylgeranyl diphosphate as a pancreatic cancer tumor growth dependency.Mol Metab. 2024 Jul;85:101964. doi: 10.1016/j.molmet.2024.101964. Epub 2024 May 31. Mol Metab. 2024. PMID: 38823776 Free PMC article.
References
-
- Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13:27–29. - PubMed
-
- Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–777. - PubMed
-
- Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279:2610–2623. - PubMed
-
- Li J, Ding S, Habib N, Fermor B, Wood C, Gilmour R. Partial characterization of a cDNA for human stearoyl-CoA desaturase and changes in its mRNA expression in some normal and malignant tissues. Int J Cancer. 1994;57:348–352. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
