

Estrogen amelioration of Aβ-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERβ, AKAP and Drp1
- PMID: 25964165
- PMCID: PMC4464937
- DOI: 10.1016/j.brainres.2015.04.059
Estrogen amelioration of Aβ-induced defects in mitochondria is mediated by mitochondrial signaling pathway involving ERβ, AKAP and Drp1
Abstract
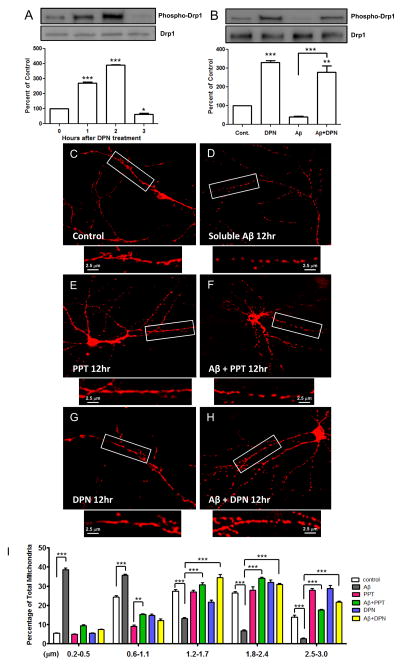
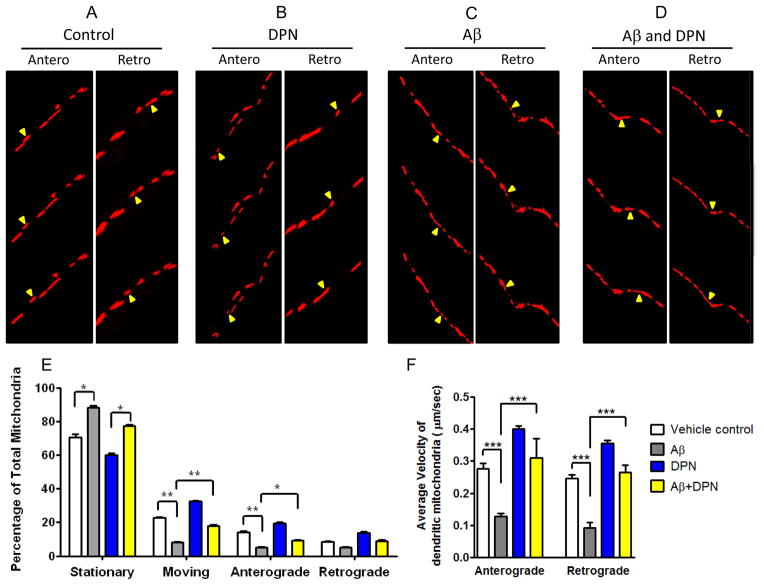
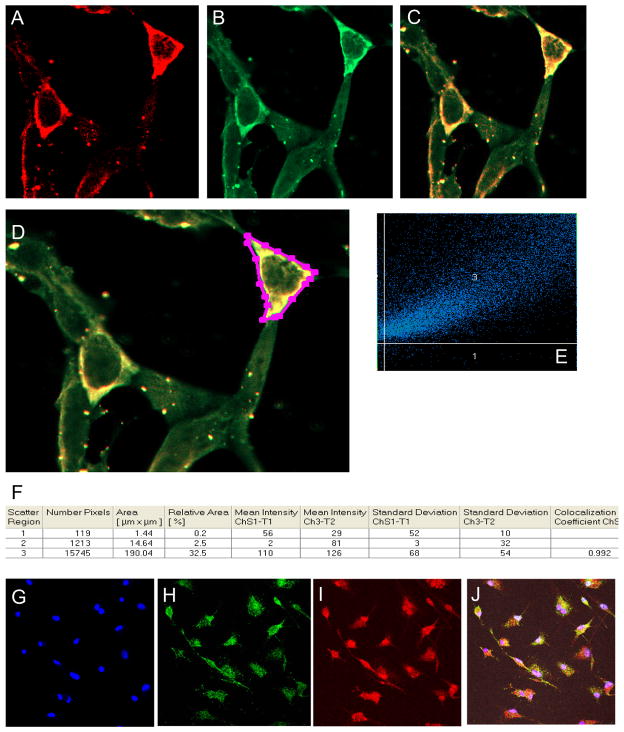
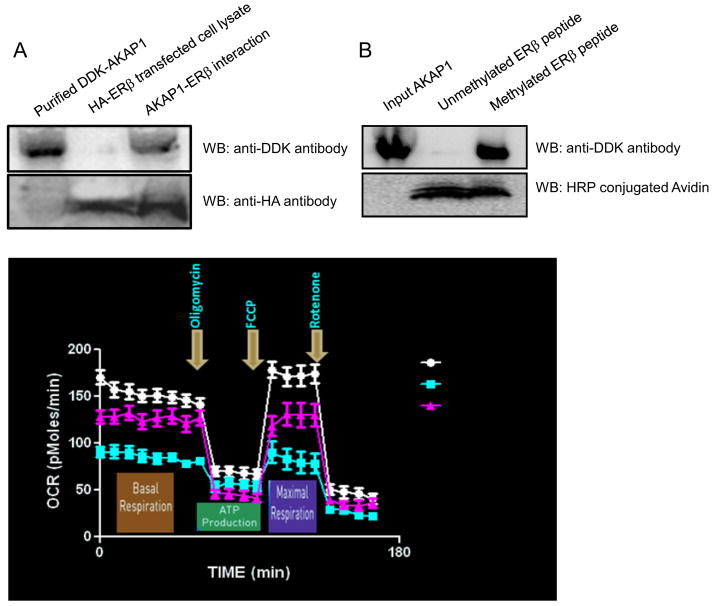
Perturbations in dynamic properties of mitochondria including fission, fusion, and movement lead to disruption of energy supply to synapses contributing to neuropathology and cognitive dysfunction in Alzheimer׳s disease (AD). The molecular mechanisms underlying these defects are still unclear. Previously, we have shown that ERβ is localized in the mitochondria and ERβ knock down disrupts mitochondrial functions. Because a selective ERβ modulator (DPN) can activate PKA, and localized PKA signaling in the mitochondrial membrane regulates mitochondrial structure and functions, we reasoned that ERβ signaling in the mitochondrial membrane rescues many of the mitochondrial defects caused by soluble Aβ oligomer. We now report that DPN treatment in primary hippocampal neurons attenuates soluble Aβ-oligomer induced dendritic mitochondrial fission and reduced mobility. Additionally, Aβ treatment reduced the respiratory reserve capacity of hippocampal neuron and inhibited phosphorylation of Drp1 at its PKA site, which induces excessive mitochondrial fission, and DPN treatment ameliorates these inhibitions. Finally, we discovered a direct interaction of ERβ with a mitochondrial resident protein AKAP1, which induces the PKA-mediated local signaling pathway involved in increased oxidative phosphorylation and inhibition of mitochondrial fission. Taken together, our findings highlight the possibility that ERβ signaling pathway may be a useful mitochondria-directed therapeutic target for AD.
Keywords: AKAP1; Alzheimer׳s disease; Estrogen receptor β; Mitochondrial Fission And Fusion; Mitochondrial Movement; PKA.
Copyright © 2015 Elsevier B.V. All rights reserved.
Conflict of interest statement
The Authors declare no conflict of interest.
Figures
Similar articles
-
Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis.Biochim Biophys Acta. 2016 Nov;1863(11):2820-2834. doi: 10.1016/j.bbamcr.2016.09.003. Epub 2016 Sep 4. Biochim Biophys Acta. 2016. PMID: 27599716
-
Mitochondrial PKA Is Neuroprotective in a Cell Culture Model of Alzheimer's Disease.Mol Neurobiol. 2021 Jul;58(7):3071-3083. doi: 10.1007/s12035-021-02333-w. Epub 2021 Feb 23. Mol Neurobiol. 2021. PMID: 33624140 Free PMC article.
-
PKA/AKAP1 and PP2A/Bβ2 regulate neuronal morphogenesis via Drp1 phosphorylation and mitochondrial bioenergetics.J Neurosci. 2011 Nov 2;31(44):15716-26. doi: 10.1523/JNEUROSCI.3159-11.2011. J Neurosci. 2011. PMID: 22049414 Free PMC article.
-
Estrogen receptor-β in mitochondria: implications for mitochondrial bioenergetics and tumorigenesis.Ann N Y Acad Sci. 2015 Sep;1350:52-60. doi: 10.1111/nyas.12872. Epub 2015 Aug 24. Ann N Y Acad Sci. 2015. PMID: 26301952 Review.
-
Mitochondria: a kinase anchoring protein 1, a signaling platform for mitochondrial form and function.Int J Biochem Cell Biol. 2014 Mar;48:92-6. doi: 10.1016/j.biocel.2013.12.012. Epub 2014 Jan 8. Int J Biochem Cell Biol. 2014. PMID: 24412345 Free PMC article. Review.
Cited by
-
Synergism between PGC-1α and estrogen in the survival of endometrial cancer cells via the mitochondrial pathway.Onco Targets Ther. 2016 Jun 30;9:3963-73. doi: 10.2147/OTT.S103482. eCollection 2016. Onco Targets Ther. 2016. PMID: 27418839 Free PMC article.
-
Intermittent fasting protects against the deterioration of cognitive function, energy metabolism and dyslipidemia in Alzheimer's disease-induced estrogen deficient rats.Exp Biol Med (Maywood). 2018 Feb;243(4):334-343. doi: 10.1177/1535370217751610. Epub 2018 Jan 7. Exp Biol Med (Maywood). 2018. PMID: 29307281 Free PMC article.
-
Effects of Female Sex Steroids Administration on Pathophysiologic Mechanisms in Traumatic Brain Injury.Transl Stroke Res. 2018 Aug;9(4):393-416. doi: 10.1007/s12975-017-0588-5. Epub 2017 Nov 19. Transl Stroke Res. 2018. PMID: 29151229 Review.
-
Estrogen deficiency exacerbates learning and memory deficits associated with glucose metabolism disorder in APP/PS1 double transgenic female mice.Genes Dis. 2021 Feb 16;9(5):1315-1331. doi: 10.1016/j.gendis.2021.01.007. eCollection 2022 Sep. Genes Dis. 2021. PMID: 35873026 Free PMC article.
-
Neurotoxic β-amyloid oligomers cause mitochondrial dysfunction-the trigger for PANoptosis in neurons.Front Aging Neurosci. 2024 May 14;16:1400544. doi: 10.3389/fnagi.2024.1400544. eCollection 2024. Front Aging Neurosci. 2024. PMID: 38808033 Free PMC article. Review.
References
-
- Attwell D, Gibb A. Neuroenergetics and the kinetic design of excitatory synapses. Nat Rev Neurosci. 2005;6:841–849. - PubMed
-
- Barghorn S, Nimmrich V, Stribinger A, Krantz C, Keller P, Janson B, Bahr M, Schmidt M, Bitner RS, Harlan J, Barlow E, Ebert U, Hilen H. Globular amyloid β-peptide1-42 oligomer-a homogenous and stable neuropathological protein in Alzheimer’s disease. J Neurochem. 2005;95:834–847. - PubMed
-
- Cardone L, Carlucci A, Affaitati A, Livigni A, DeCristofaro T, Garbi C, Varrone S, Ullrich A, Gottesman ME, Avvedimento EV, Feliciello A. Mitochondrial AKAP121 binds and targets protein tyrosine phosphatase D1, a novel positive regulator of src signaling. Mol Cell Biol. 2004;24:4613–26. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
