

Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease
- PMID: 25943890
- PMCID: PMC4470809
- DOI: 10.1007/s00401-015-1436-x
Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease
Abstract
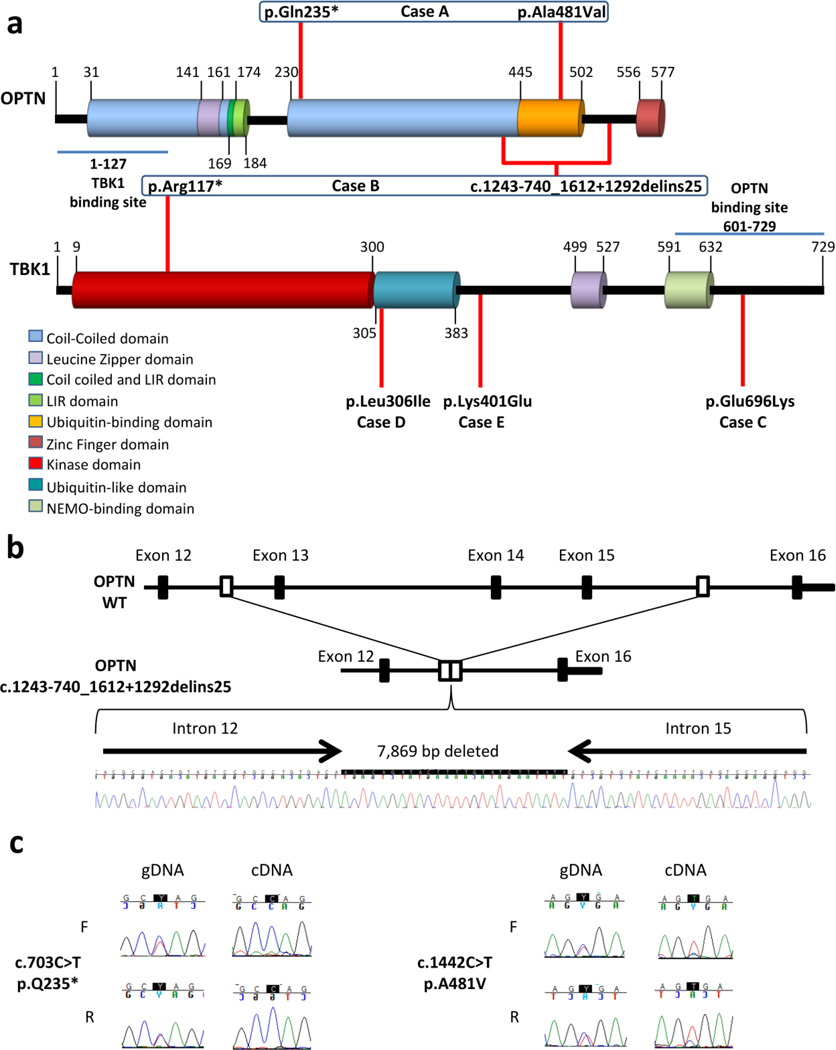
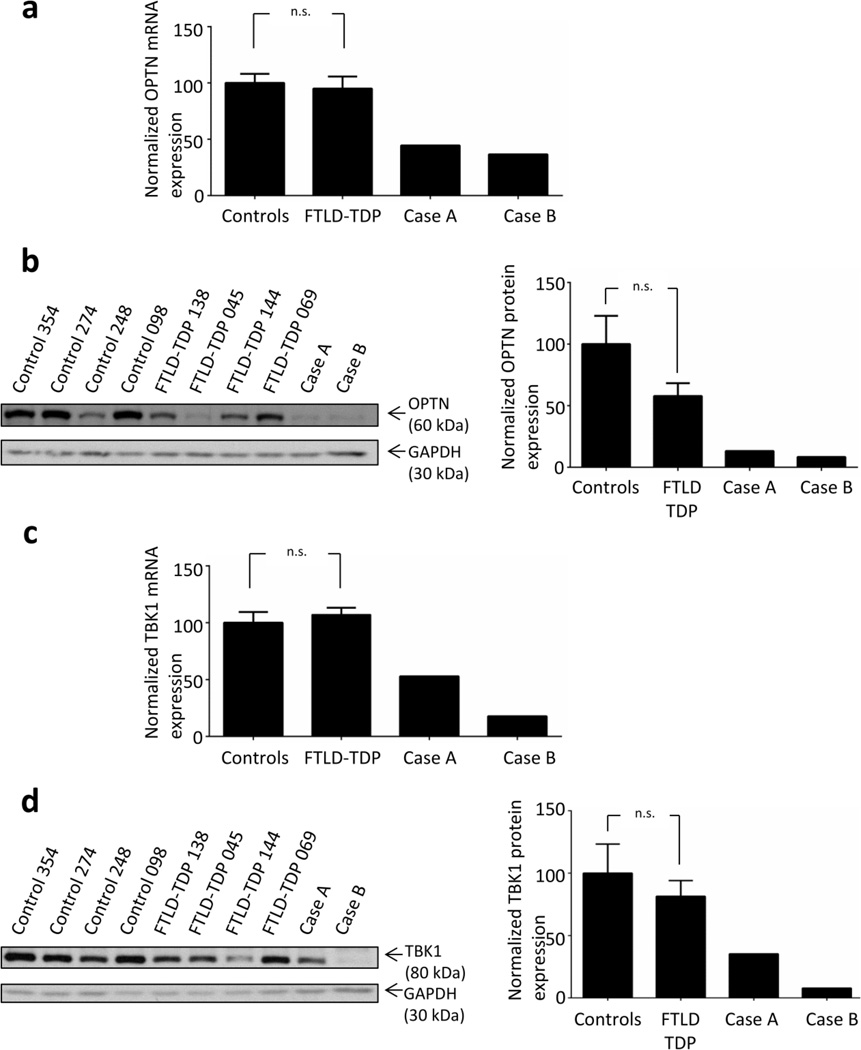
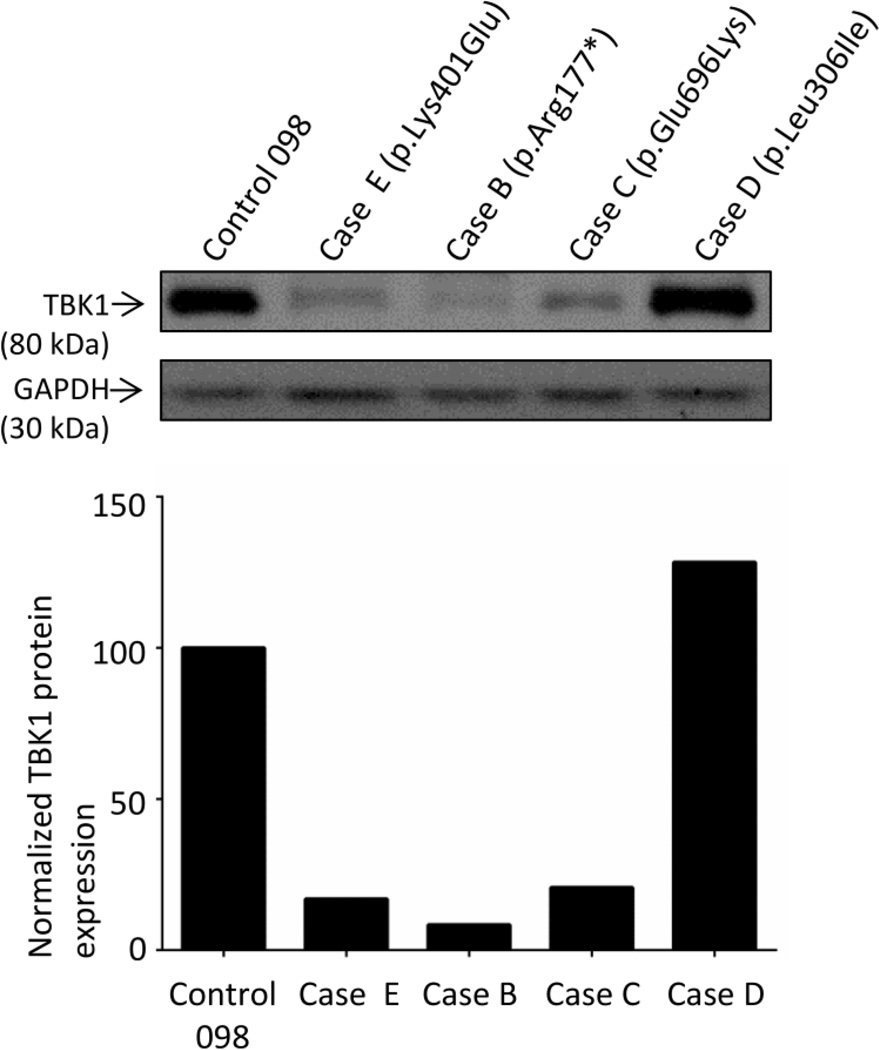
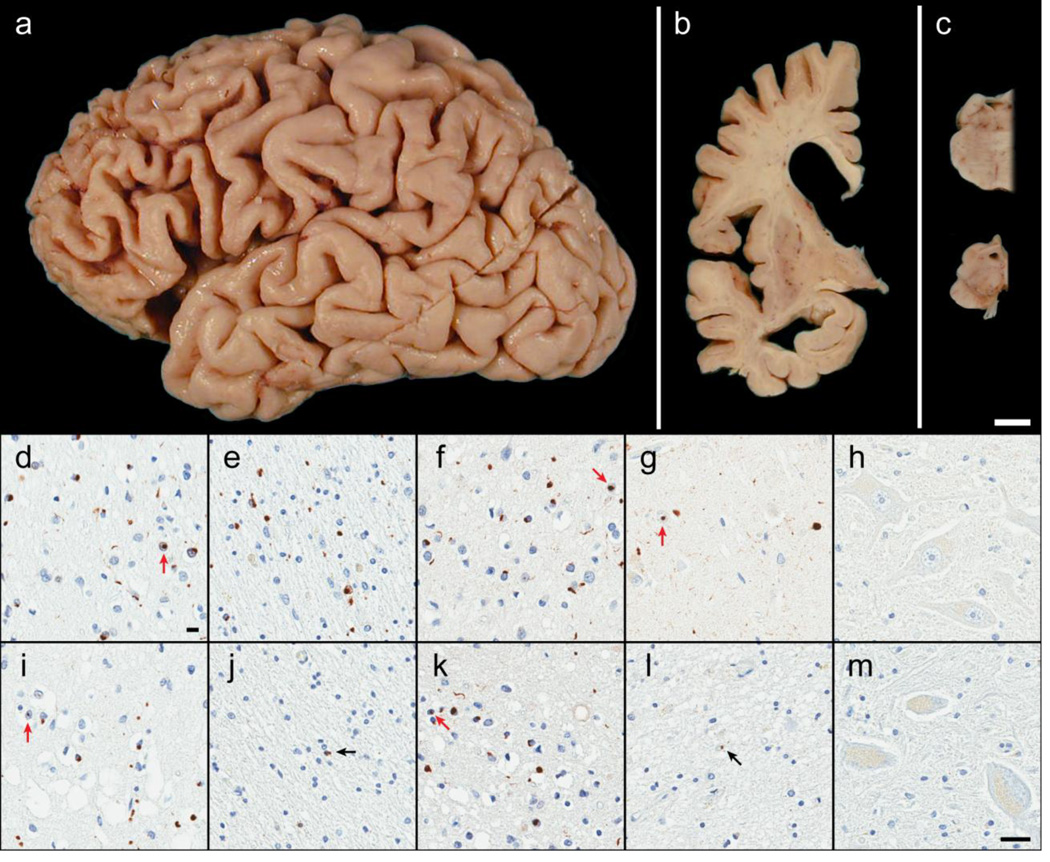
Frontotemporal lobar degeneration with TAR DNA-binding protein 43 inclusions (FTLD-TDP) is the most common pathology associated with frontotemporal dementia (FTD). Repeat expansions in chromosome 9 open reading frame 72 (C9ORF72) and mutations in progranulin (GRN) are the major known genetic causes of FTLD-TDP; however, the genetic etiology in the majority of FTLD-TDP remains unexplained. In this study, we performed whole-genome sequencing in 104 pathologically confirmed FTLD-TDP patients from the Mayo Clinic brain bank negative for C9ORF72 and GRN mutations and report on the contribution of rare single nucleotide and copy number variants in 21 known neurodegenerative disease genes. Interestingly, we identified 5 patients (4.8 %) with variants in optineurin (OPTN) and TANK-binding kinase 1 (TBK1) that are predicted to be highly pathogenic, including two double mutants. Case A was a compound heterozygote for mutations in OPTN, carrying the p.Q235* nonsense and p.A481V missense mutation in trans, while case B carried a deletion of OPTN exons 13-15 (p.Gly538Glufs*27) and a loss-of-function mutation (p.Arg117*) in TBK1. Cases C-E carried heterozygous missense mutations in TBK1, including the p.Glu696Lys mutation which was previously reported in two amyotrophic lateral sclerosis (ALS) patients and is located in the OPTN binding domain. Quantitative mRNA expression and protein analysis in cerebellar tissue showed a striking reduction of OPTN and/or TBK1 expression in 4 out of 5 patients supporting pathogenicity in these specific patients and suggesting a loss-of-function disease mechanism. Importantly, neuropathologic examination showed FTLD-TDP type A in the absence of motor neuron disease in 3 pathogenic mutation carriers. In conclusion, we highlight TBK1 as an important cause of pure FTLD-TDP, identify the first OPTN mutations in FTLD-TDP, and suggest a potential oligogenic basis for at least a subset of FTLD-TDP patients. Our data further add to the growing body of evidence linking ALS and FTD and suggest a key role for the OPTN/TBK1 pathway in these diseases.
Conflict of interest statement
Disclosure of potential conflicts of interest:
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders.Acta Neuropathol. 2011 Apr;121(4):519-27. doi: 10.1007/s00401-011-0813-3. Epub 2011 Mar 1. Acta Neuropathol. 2011. PMID: 21360076
-
Genome-wide analyses as part of the international FTLD-TDP whole-genome sequencing consortium reveals novel disease risk factors and increases support for immune dysfunction in FTLD.Acta Neuropathol. 2019 Jun;137(6):879-899. doi: 10.1007/s00401-019-01962-9. Epub 2019 Feb 9. Acta Neuropathol. 2019. PMID: 30739198 Free PMC article.
-
Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis.Neurobiol Dis. 2022 Nov;174:105859. doi: 10.1016/j.nbd.2022.105859. Epub 2022 Sep 13. Neurobiol Dis. 2022. PMID: 36113750 Review.
-
Clinical features of TBK1 carriers compared with C9orf72, GRN and non-mutation carriers in a Belgian cohort.Brain. 2016 Feb;139(Pt 2):452-67. doi: 10.1093/brain/awv358. Epub 2015 Dec 15. Brain. 2016. PMID: 26674655 Free PMC article.
-
Frontotemporal lobar degeneration and amyotrophic lateral sclerosis: molecular similarities and differences.Rev Neurol (Paris). 2013 Oct;169(10):793-8. doi: 10.1016/j.neurol.2013.07.019. Epub 2013 Sep 5. Rev Neurol (Paris). 2013. PMID: 24011641 Review.
Cited by
-
Genetic spectrum features and diagnostic accuracy of four plasma biomarkers in 248 Chinese patients with frontotemporal dementia.Alzheimers Dement. 2024 Oct;20(10):7281-7295. doi: 10.1002/alz.14215. Epub 2024 Sep 10. Alzheimers Dement. 2024. PMID: 39254359 Free PMC article.
-
Mitochondria-associated membrane collapse impairs TBK1-mediated proteostatic stress response in ALS.Proc Natl Acad Sci U S A. 2023 Nov 21;120(47):e2315347120. doi: 10.1073/pnas.2315347120. Epub 2023 Nov 15. Proc Natl Acad Sci U S A. 2023. PMID: 37967220 Free PMC article.
-
Network-based approach for targeting human kinases commonly associated with amyotrophic lateral sclerosis and cancer.Front Mol Neurosci. 2022 Dec 16;15:1023286. doi: 10.3389/fnmol.2022.1023286. eCollection 2022. Front Mol Neurosci. 2022. PMID: 36590916 Free PMC article.
-
Patients with sporadic FTLD exhibit similar increases in lysosomal proteins and storage material as patients with FTD due to GRN mutations.Acta Neuropathol Commun. 2023 Apr 28;11(1):70. doi: 10.1186/s40478-023-01571-4. Acta Neuropathol Commun. 2023. PMID: 37118844 Free PMC article.
-
Recent advances in amyotrophic lateral sclerosis.J Neurol. 2016 Jun;263(6):1241-54. doi: 10.1007/s00415-016-8091-6. Epub 2016 Mar 30. J Neurol. 2016. PMID: 27025851 Free PMC article. Review.
References
-
- Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. - PubMed
-
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. - PubMed
-
- Arai T, Nonaka T, Hasegawa M, Akiyama H, Yoshida M, Hashizume Y, Tsuchiya K, Oda T, Ikeda K. Neuronal and glial inclusions in frontotemporal dementia with or without motor neuron disease are immunopositive for p62. Neurosci Lett. 2003;342:41–44. - PubMed
-
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. - PubMed
-
- Belzil VV, Daoud H, Desjarlais A, Bouchard JP, Dupre N, Camu W, Dion PA, Rouleau GA. Analysis of OPTN as a causative gene for amyotrophic lateral sclerosis. Neurobiology of aging. 2011;32:555, e513–e554. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
