

A simple biophysical model emulates budding yeast chromosome condensation
- PMID: 25922992
- PMCID: PMC4413874
- DOI: 10.7554/eLife.05565
A simple biophysical model emulates budding yeast chromosome condensation
Abstract
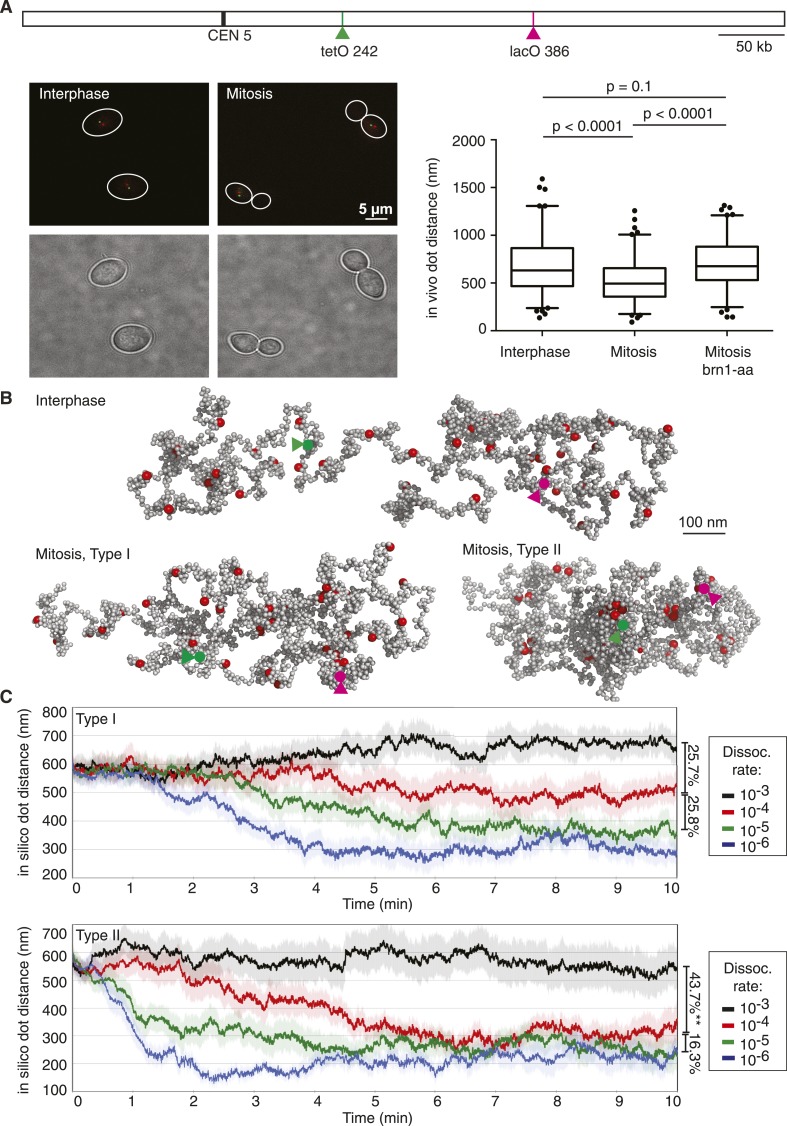
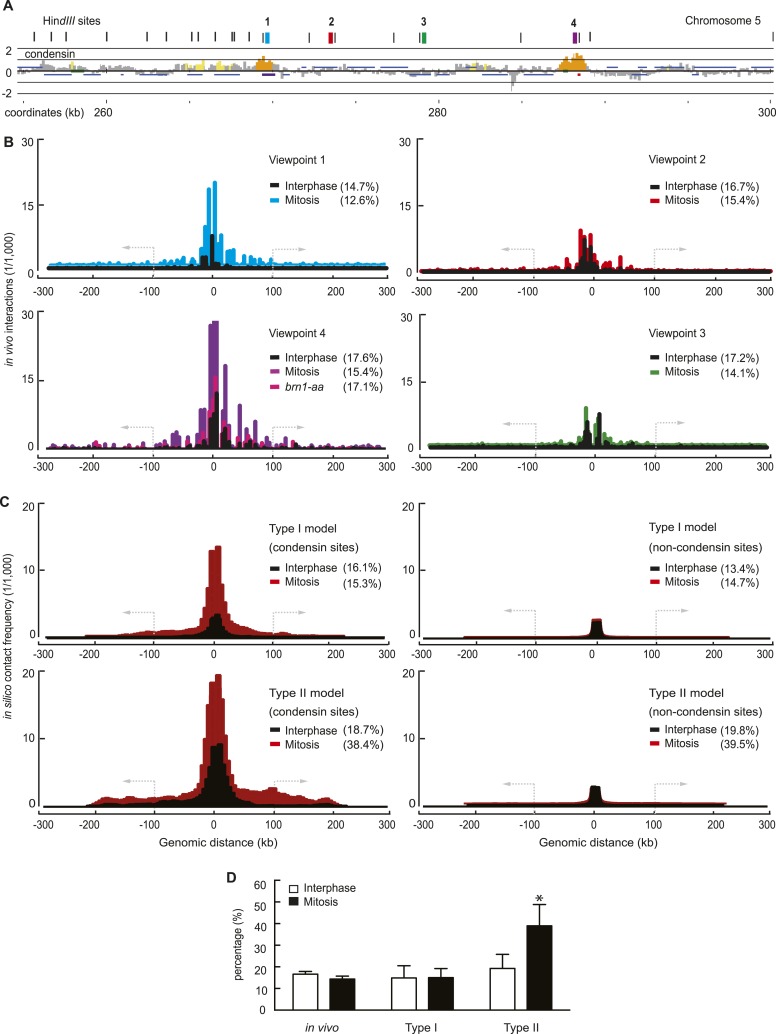
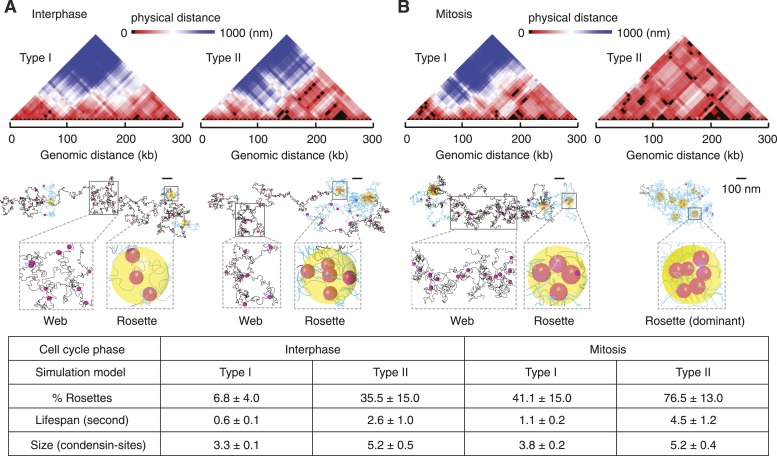
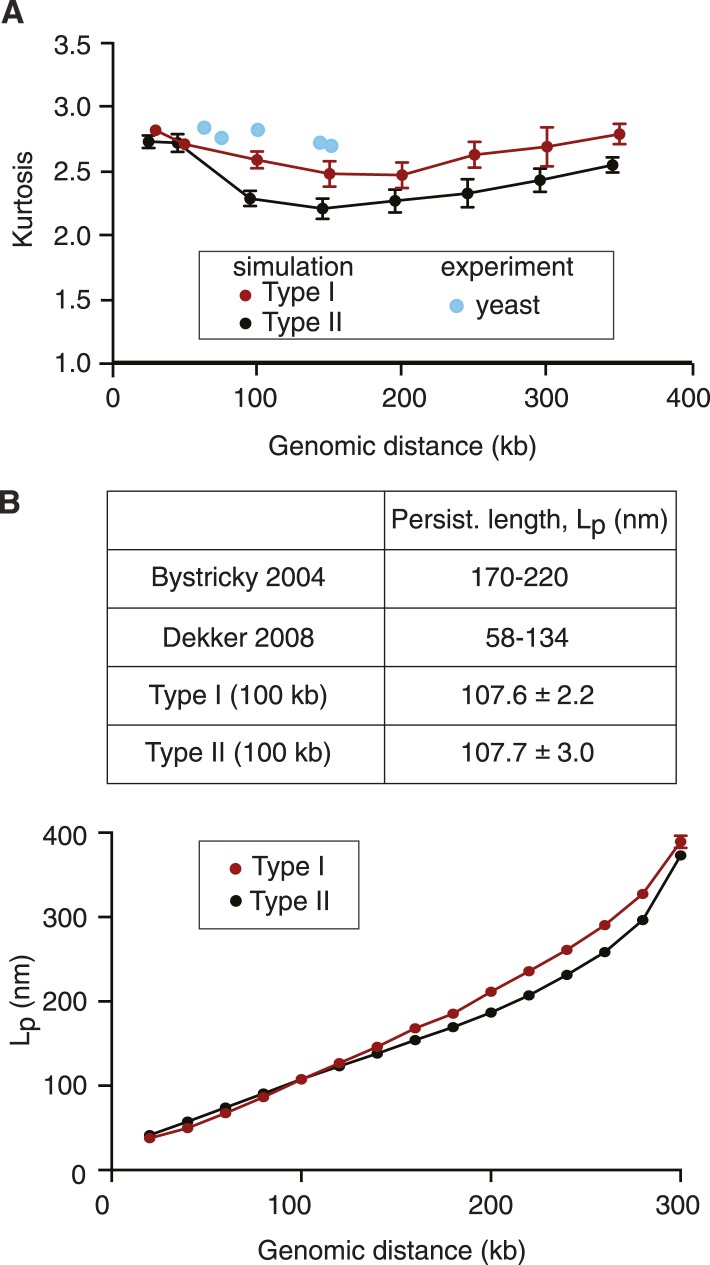
Mitotic chromosomes were one of the first cell biological structures to be described, yet their molecular architecture remains poorly understood. We have devised a simple biophysical model of a 300 kb-long nucleosome chain, the size of a budding yeast chromosome, constrained by interactions between binding sites of the chromosomal condensin complex, a key component of interphase and mitotic chromosomes. Comparisons of computational and experimental (4C) interaction maps, and other biophysical features, allow us to predict a mode of condensin action. Stochastic condensin-mediated pairwise interactions along the nucleosome chain generate native-like chromosome features and recapitulate chromosome compaction and individualization during mitotic condensation. Higher order interactions between condensin binding sites explain the data less well. Our results suggest that basic assumptions about chromatin behavior go a long way to explain chromosome architecture and are able to generate a molecular model of what the inside of a chromosome is likely to look like.
Keywords: S. cerevisiae; chromosome architecture; chromosomes; computational biology; condensin; genes; mitosis; systems biology.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures





Similar articles
-
[Functional characteristics of the individual genomic condensin binding sites of Saccharomyces cerevisiae using minichromosome mitotic segregation stability model].Tsitologiia. 2008;50(9):788-93. Tsitologiia. 2008. PMID: 18959191 Russian.
-
SMC complexes differentially compact mitotic chromosomes according to genomic context.Nat Cell Biol. 2017 Sep;19(9):1071-1080. doi: 10.1038/ncb3594. Epub 2017 Aug 21. Nat Cell Biol. 2017. PMID: 28825700 Free PMC article.
-
Cdc48/VCP Promotes Chromosome Morphogenesis by Releasing Condensin from Self-Entrapment in Chromatin.Mol Cell. 2018 Feb 15;69(4):664-676.e5. doi: 10.1016/j.molcel.2018.01.030. Mol Cell. 2018. PMID: 29452641
-
Three-step model for condensin activation during mitotic chromosome condensation.Cell Cycle. 2010 Aug 15;9(16):3243-55. doi: 10.4161/cc.9.16.12620. Epub 2010 Aug 7. Cell Cycle. 2010. PMID: 20703077 Review.
-
Condensins and cohesins - one of these things is not like the other!J Cell Sci. 2019 Feb 7;132(3):jcs220491. doi: 10.1242/jcs.220491. J Cell Sci. 2019. PMID: 30733374 Free PMC article. Review.
Cited by
-
Large domains of heterochromatin direct the formation of short mitotic chromosome loops.Elife. 2020 Sep 11;9:e57212. doi: 10.7554/eLife.57212. Elife. 2020. PMID: 32915140 Free PMC article.
-
Agent-based modeling of nuclear chromosome ensemble identifies determinants of homolog pairing during meiosis.bioRxiv [Preprint]. 2024 Jan 13:2023.08.09.552574. doi: 10.1101/2023.08.09.552574. bioRxiv. 2024. Update in: PLoS Comput Biol. 2024 May 13;20(5):e1011416. doi: 10.1371/journal.pcbi.1011416. PMID: 38260664 Free PMC article. Updated. Preprint.
-
An extrinsic motor directs chromatin loop formation by cohesin.EMBO J. 2024 Oct;43(19):4173-4196. doi: 10.1038/s44318-024-00202-5. Epub 2024 Aug 19. EMBO J. 2024. PMID: 39160275 Free PMC article.
-
Organization of the Escherichia coli Chromosome by a MukBEF Axial Core.Mol Cell. 2020 Apr 16;78(2):250-260.e5. doi: 10.1016/j.molcel.2020.02.003. Epub 2020 Feb 24. Mol Cell. 2020. PMID: 32097603 Free PMC article.
-
Involvement of condensin-directed gene associations in the organization and regulation of chromosome territories during the cell cycle.Nucleic Acids Res. 2016 May 5;44(8):3618-28. doi: 10.1093/nar/gkv1502. Epub 2015 Dec 23. Nucleic Acids Res. 2016. PMID: 26704981 Free PMC article.
References
-
- Balanda KP, MacGillivray HL. Kurtosis: a critical review. Americ Statistician. 1988;42:111–119.
-
- Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proceedings of the National Academy of Sciences of USA. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
