

Studies on the Contribution of Human Cytomegalovirus UL21a and UL97 to Viral Growth and Inactivation of the Anaphase-Promoting Complex/Cyclosome (APC/C) E3 Ubiquitin Ligase Reveal a Unique Cellular Mechanism for Downmodulation of the APC/C Subunits APC1, APC4, and APC5
- PMID: 25903336
- PMCID: PMC4468507
- DOI: 10.1128/JVI.00403-15
Studies on the Contribution of Human Cytomegalovirus UL21a and UL97 to Viral Growth and Inactivation of the Anaphase-Promoting Complex/Cyclosome (APC/C) E3 Ubiquitin Ligase Reveal a Unique Cellular Mechanism for Downmodulation of the APC/C Subunits APC1, APC4, and APC5
Abstract
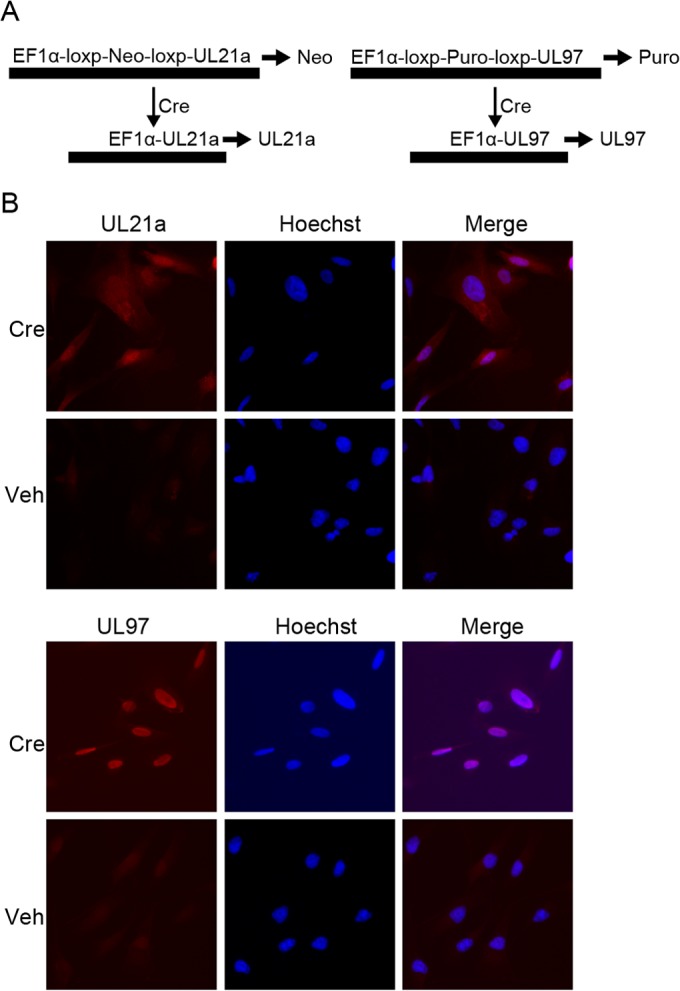
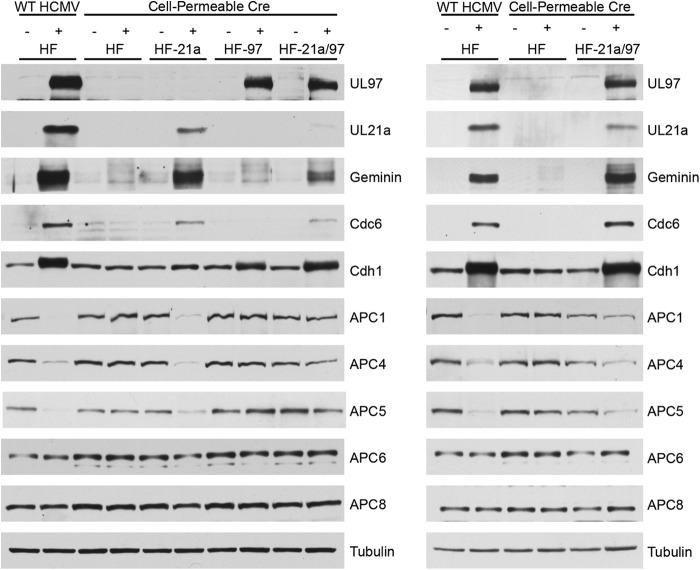
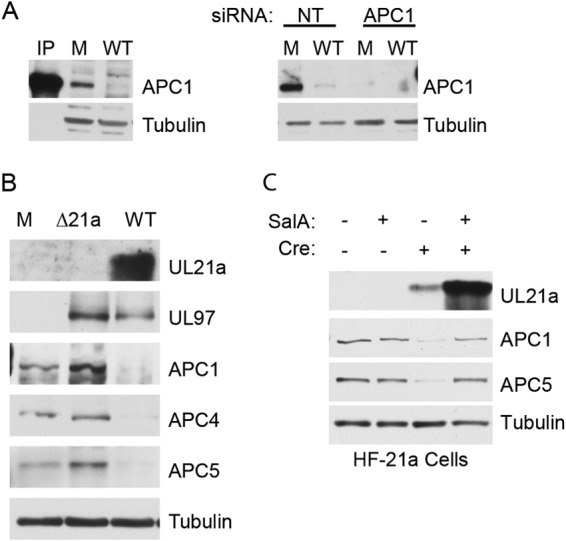
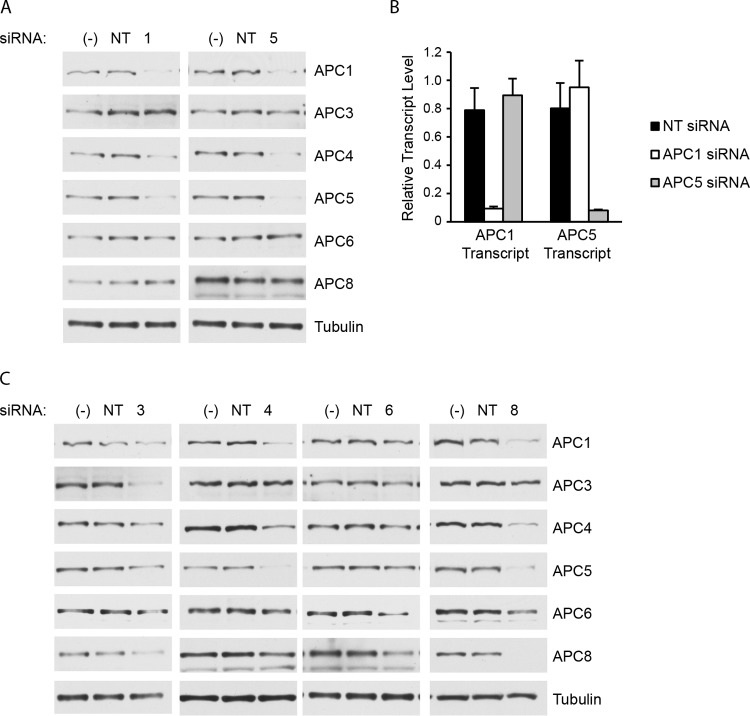
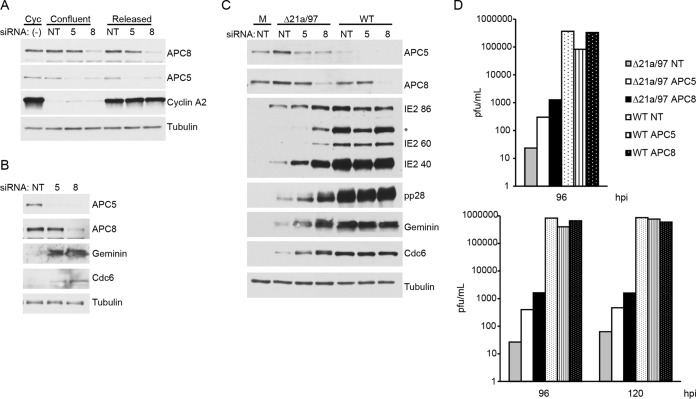
Human cytomegalovirus (HCMV) deregulates the cell cycle by several means, including inactivation of the anaphase-promoting complex/cyclosome (APC/C) E3 ubiquitin ligase. Viral proteins UL97 and UL21a, respectively, affect the APC/C by phosphorylation of APC/C coactivator Cdh1 and by inducing the degradation of subunits APC4 and APC5, which along with APC1 form the APC/C platform subcomplex. The aim of this study was to further characterize the mechanism of APC/C inactivation and define the relative contributions of UL21a and UL97 to APC/C substrate accumulation and to viral growth. We show that in uninfected cells, UL21a but not UL97 can disrupt APC/C function, leading to the accumulation of substrates. We find that UL21a is necessary and sufficient to induce the degradation of APC1, in addition to the previously reported APC4 and APC5. We also demonstrate that there is a previously unreported cellular mechanism for a specific decrease in the levels of all three platform subunits, APC1, APC4, and APC5, upon the depletion of any one of these subunits or of subunit APC8. Finally, we show that at a low multiplicity of infection, either UL97 or UL21a can partially complement a growth-defective mutant virus lacking both UL21a and UL97, with significantly greater benefit afforded by the expression of both proteins. This double mutant also can be partially rescued by inactivation of the APC/C using small interfering RNAs against specific subunits. These results further our understanding of HCMV's interaction with the cell cycle machinery and reveal a new cellular pattern of APC/C subunit downmodulation.
Importance: HCMV lytic infection subverts the host cell cycle machinery in multiple ways. A major effect is inactivation of the APC/C, which plays a central role in the control of cell cycle progression. This study provides further insight into the mechanism of inactivation. We discovered that the APC1 subunit, which along with APC4 and APC5 form the platform subcomplex of the APC/C, is an additional target of the degradation induced by HCMV protein UL21a. This study also shows for the first time that there is a unique cellular process in uninfected cells whereby depletion of APC1, APC4, APC5, or APC8 recapitulates the pattern of HCMV-mediated APC/C subunit degradation.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
Inactivation and disassembly of the anaphase-promoting complex during human cytomegalovirus infection is associated with degradation of the APC5 and APC4 subunits and does not require UL97-mediated phosphorylation of Cdh1.J Virol. 2010 Oct;84(20):10832-43. doi: 10.1128/JVI.01260-10. Epub 2010 Aug 4. J Virol. 2010. PMID: 20686030 Free PMC article.
-
TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1.Curr Biol. 2003 Sep 2;13(17):1459-68. doi: 10.1016/s0960-9822(03)00581-5. Curr Biol. 2003. PMID: 12956947
-
Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a.PLoS Pathog. 2012;8(7):e1002789. doi: 10.1371/journal.ppat.1002789. Epub 2012 Jul 5. PLoS Pathog. 2012. PMID: 22792066 Free PMC article.
-
Control of metaphase-anaphase progression by proteolysis: cyclosome function regulated by the protein kinase A pathway, ubiquitination and localization.Philos Trans R Soc Lond B Biol Sci. 1999 Sep 29;354(1389):1559-69; discussion 1569-70. doi: 10.1098/rstb.1999.0499. Philos Trans R Soc Lond B Biol Sci. 1999. PMID: 10582241 Free PMC article. Review.
-
Control the host cell cycle: viral regulation of the anaphase-promoting complex.J Virol. 2013 Aug;87(16):8818-25. doi: 10.1128/JVI.00088-13. Epub 2013 Jun 12. J Virol. 2013. PMID: 23760246 Free PMC article. Review.
Cited by
-
Understanding the Cytomegalovirus Cyclin-Dependent Kinase Ortholog pUL97 as a Multifaceted Regulator and an Antiviral Drug Target.Cells. 2024 Aug 13;13(16):1338. doi: 10.3390/cells13161338. Cells. 2024. PMID: 39195228 Free PMC article. Review.
-
Human Immunodeficiency Virus Type 1 Vpr Mediates Degradation of APC1, a Scaffolding Component of the Anaphase-Promoting Complex/Cyclosome.J Virol. 2021 Jul 12;95(15):e0097120. doi: 10.1128/JVI.00971-20. Epub 2021 Jul 12. J Virol. 2021. PMID: 34011540 Free PMC article.
-
Network mechanisms and dysfunction within an integrated computational model of progression through mitosis in the human cell cycle.PLoS Comput Biol. 2020 Apr 6;16(4):e1007733. doi: 10.1371/journal.pcbi.1007733. eCollection 2020 Apr. PLoS Comput Biol. 2020. PMID: 32251461 Free PMC article.
-
Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor.Proc Natl Acad Sci U S A. 2020 Aug 4;117(31):18771-18779. doi: 10.1073/pnas.2001887117. Epub 2020 Jul 20. Proc Natl Acad Sci U S A. 2020. PMID: 32690704 Free PMC article.
-
Virus-host protein interactions as footprints of human cytomegalovirus replication.Curr Opin Virol. 2022 Feb;52:135-147. doi: 10.1016/j.coviro.2021.11.016. Epub 2021 Dec 16. Curr Opin Virol. 2022. PMID: 34923282 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
