

Calibrating genomic and allelic coverage bias in single-cell sequencing
- PMID: 25879913
- PMCID: PMC4922254
- DOI: 10.1038/ncomms7822
Calibrating genomic and allelic coverage bias in single-cell sequencing
Abstract
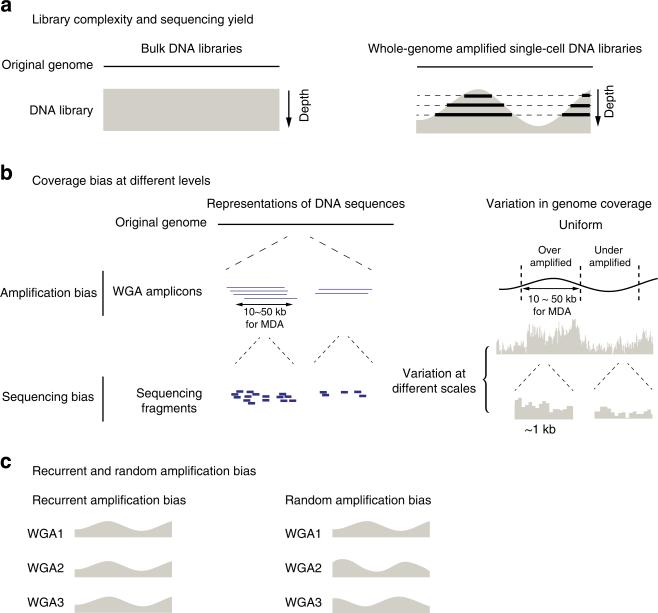
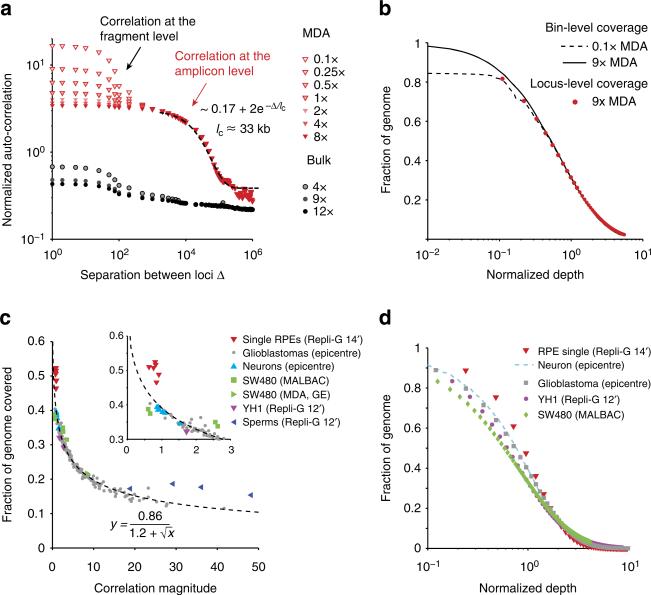
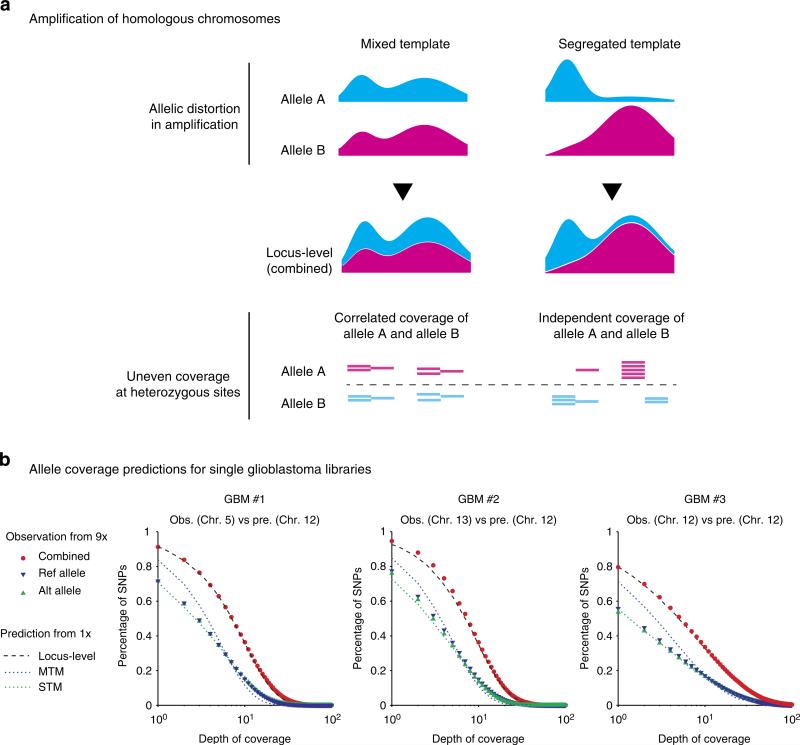
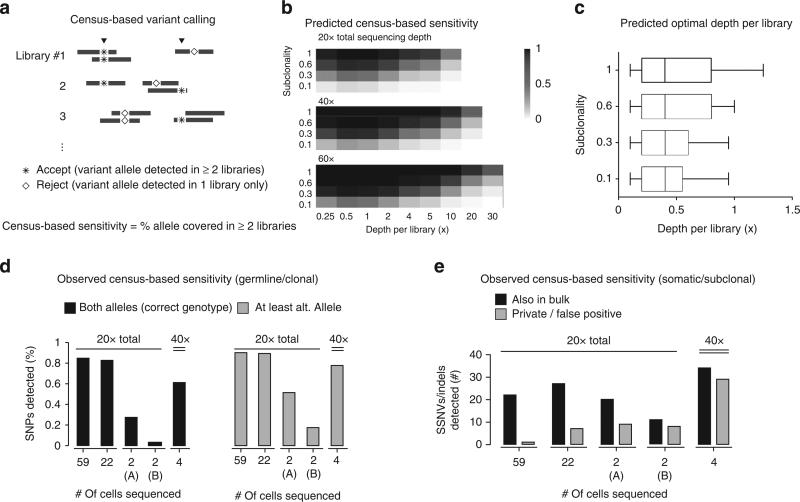
Artifacts introduced in whole-genome amplification (WGA) make it difficult to derive accurate genomic information from single-cell genomes and require different analytical strategies from bulk genome analysis. Here, we describe statistical methods to quantitatively assess the amplification bias resulting from whole-genome amplification of single-cell genomic DNA. Analysis of single-cell DNA libraries generated by different technologies revealed universal features of the genome coverage bias predominantly generated at the amplicon level (1-10 kb). The magnitude of coverage bias can be accurately calibrated from low-pass sequencing (∼0.1 × ) to predict the depth-of-coverage yield of single-cell DNA libraries sequenced at arbitrary depths. We further provide a benchmark comparison of single-cell libraries generated by multi-strand displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC). Finally, we develop statistical models to calibrate allelic bias in single-cell whole-genome amplification and demonstrate a census-based strategy for efficient and accurate variant detection from low-input biopsy samples.
Figures
Similar articles
-
Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates.Proc Natl Acad Sci U S A. 2016 Jul 26;113(30):8484-9. doi: 10.1073/pnas.1520964113. Epub 2016 Jul 13. Proc Natl Acad Sci U S A. 2016. PMID: 27412862 Free PMC article.
-
Comparison of multiple displacement amplification (MDA) and multiple annealing and looping-based amplification cycles (MALBAC) in single-cell sequencing.PLoS One. 2014 Dec 8;9(12):e114520. doi: 10.1371/journal.pone.0114520. eCollection 2014. PLoS One. 2014. PMID: 25485707 Free PMC article.
-
[Comparison of different single cell whole genome amplification methods and MALBAC applications in assisted reproduction].Yi Chuan. 2018 Aug 16;40(8):620-631. doi: 10.16288/j.yczz.18-091. Yi Chuan. 2018. PMID: 30117418 Review. Chinese.
-
Precision oncology using a limited number of cells: optimization of whole genome amplification products for sequencing applications.BMC Cancer. 2017 Jul 1;17(1):457. doi: 10.1186/s12885-017-3447-6. BMC Cancer. 2017. PMID: 28666423 Free PMC article.
-
Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications.Annu Rev Genomics Hum Genet. 2015;16:79-102. doi: 10.1146/annurev-genom-090413-025352. Epub 2015 Jun 11. Annu Rev Genomics Hum Genet. 2015. PMID: 26077818 Review.
Cited by
-
Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology.Nucleic Acids Res. 2020 Feb 20;48(3):1146-1163. doi: 10.1093/nar/gkz1173. Nucleic Acids Res. 2020. PMID: 31853540 Free PMC article.
-
Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network.Science. 2017 Apr 28;356(6336):eaal1641. doi: 10.1126/science.aal1641. Epub 2017 Apr 27. Science. 2017. PMID: 28450582 Free PMC article. Review.
-
De novo detection of somatic mutations in high-throughput single-cell profiling data sets.Nat Biotechnol. 2024 May;42(5):758-767. doi: 10.1038/s41587-023-01863-z. Epub 2023 Jul 6. Nat Biotechnol. 2024. PMID: 37414936 Free PMC article.
-
Chromothripsis from DNA damage in micronuclei.Nature. 2015 Jun 11;522(7555):179-84. doi: 10.1038/nature14493. Epub 2015 May 27. Nature. 2015. PMID: 26017310 Free PMC article.
-
Hotspot Selective Preference of the Chimeric Sequences Formed in Multiple Displacement Amplification.Int J Mol Sci. 2017 Feb 24;18(3):492. doi: 10.3390/ijms18030492. Int J Mol Sci. 2017. PMID: 28245591 Free PMC article.
References
-
- Shapiro E, Biezuner T, Linnarsson S. Single-cell sequencing-based technologies will revolutionize whole-organism science. Nat. Rev. Genet. 2013;14:618–630. - PubMed
-
- Chi KR. Singled out for sequencing. Nat. Methods. 2014;11:13–17. - PubMed
-
- Hou Y, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873–885. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
