

Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization
- PMID: 25866904
- PMCID: PMC4411683
- DOI: 10.3390/v7041927
Insect-specific flaviviruses: a systematic review of their discovery, host range, mode of transmission, superinfection exclusion potential and genomic organization
Abstract
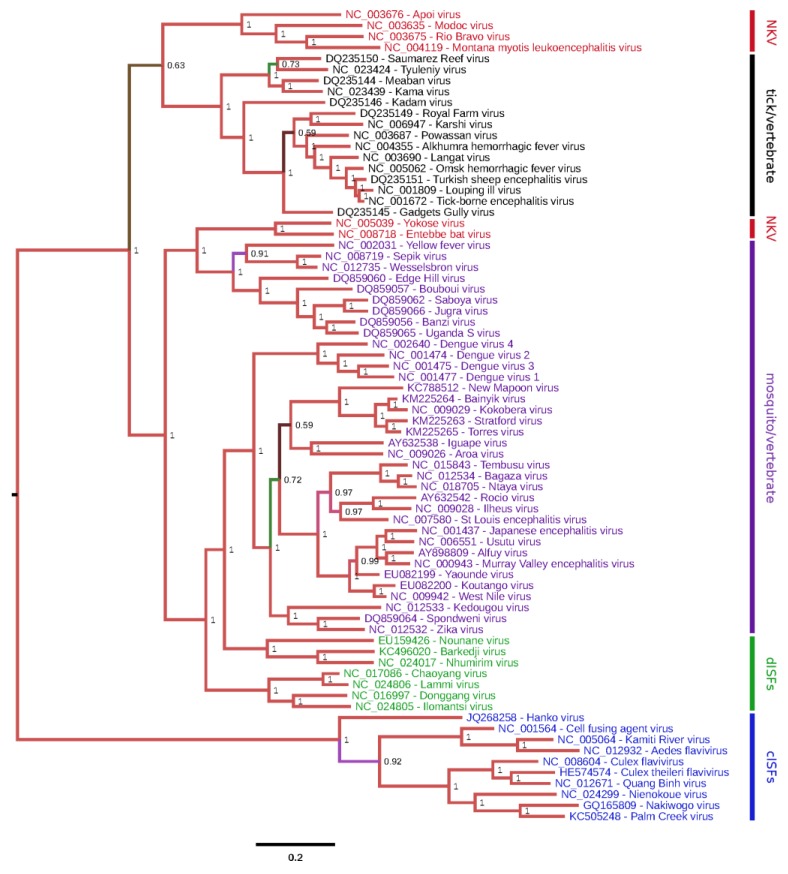
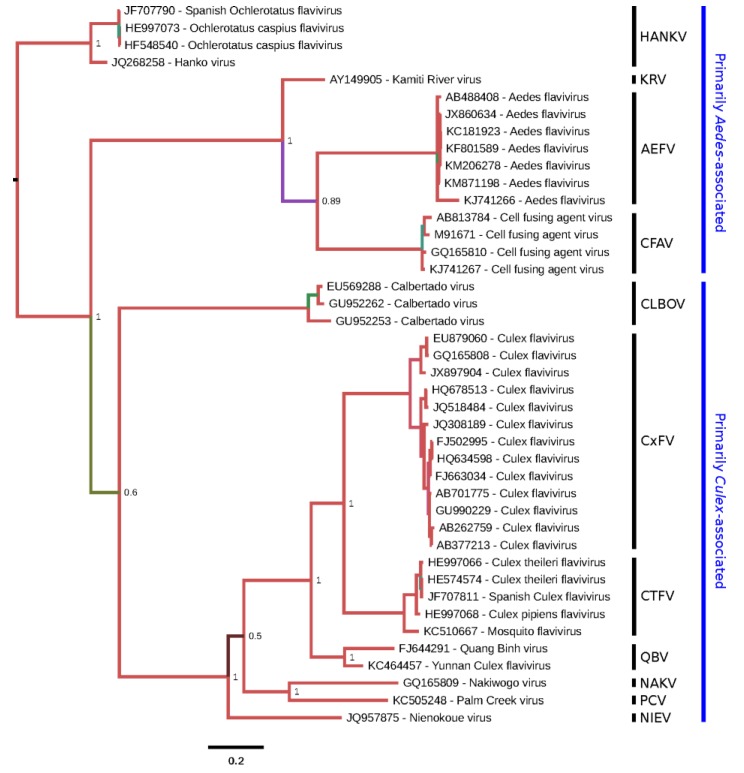
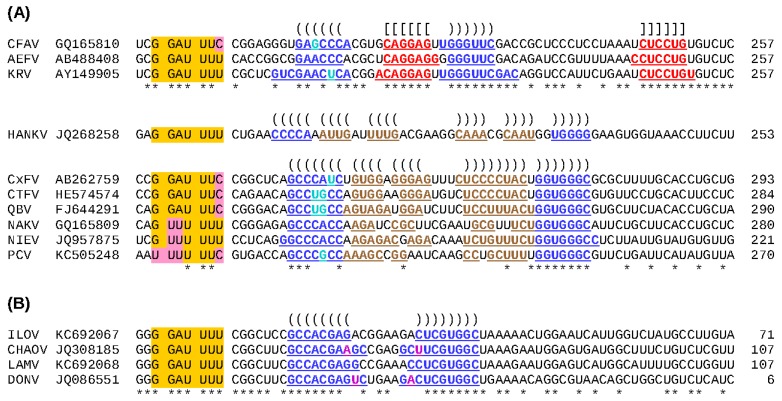
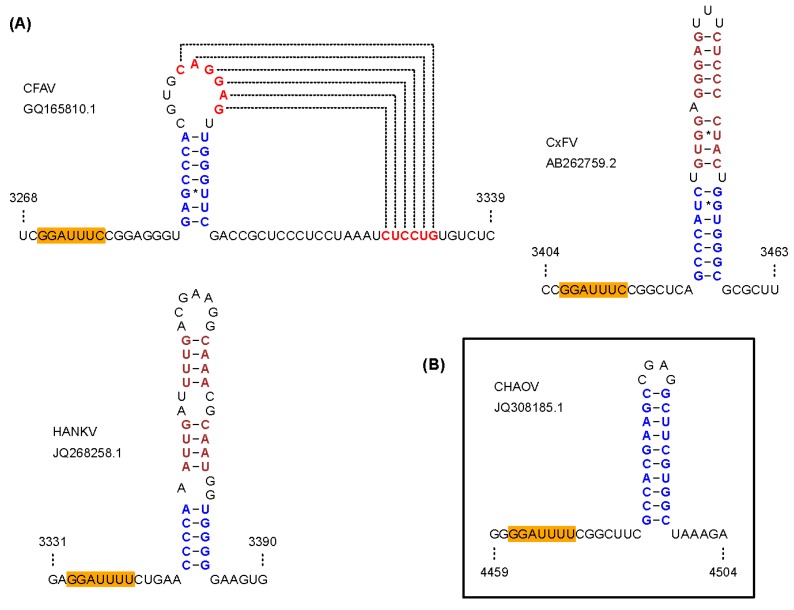
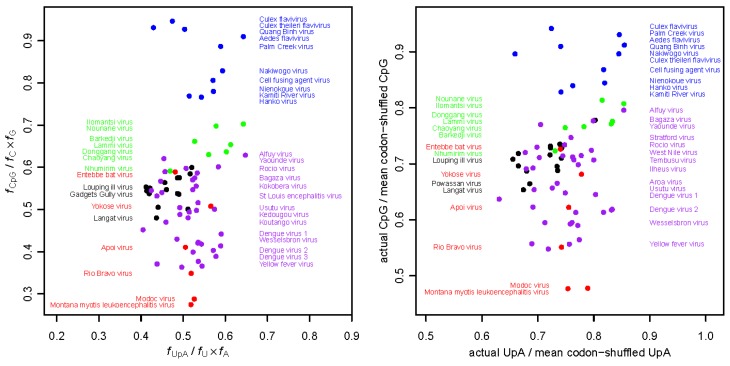
There has been a dramatic increase in the number of insect-specific flaviviruses (ISFs) discovered in the last decade. Historically, these viruses have generated limited interest due to their inability to infect vertebrate cells. This viewpoint has changed in recent years because some ISFs have been shown to enhance or suppress the replication of medically important flaviviruses in co-infected mosquito cells. Additionally, comparative studies between ISFs and medically important flaviviruses can provide a unique perspective as to why some flaviviruses possess the ability to infect and cause devastating disease in humans while others do not. ISFs have been isolated exclusively from mosquitoes in nature but the detection of ISF-like sequences in sandflies and chironomids indicates that they may also infect other dipterans. ISFs can be divided into two distinct phylogenetic groups. The first group currently consists of approximately 12 viruses and includes cell fusing agent virus, Kamiti River virus and Culex flavivirus. These viruses are phylogenetically distinct from all other known flaviviruses. The second group, which is apparently not monophyletic, currently consists of nine viruses and includes Chaoyang virus, Nounané virus and Lammi virus. These viruses phylogenetically affiliate with mosquito/vertebrate flaviviruses despite their apparent insect-restricted phenotype. This article provides a review of the discovery, host range, mode of transmission, superinfection exclusion ability and genomic organization of ISFs. This article also attempts to clarify the ISF nomenclature because some of these viruses have been assigned more than one name due to their simultaneous discoveries by independent research groups.
Figures
Similar articles
-
Antigenic Characterization of New Lineage II Insect-Specific Flaviviruses in Australian Mosquitoes and Identification of Host Restriction Factors.mSphere. 2020 Jun 17;5(3):e00095-20. doi: 10.1128/mSphere.00095-20. mSphere. 2020. PMID: 32554715 Free PMC article.
-
Insect-specific flaviviruses, a worldwide widespread group of viruses only detected in insects.Infect Genet Evol. 2016 Jun;40:381-388. doi: 10.1016/j.meegid.2015.07.032. Epub 2015 Jul 31. Infect Genet Evol. 2016. PMID: 26235844
-
A new insect-specific flavivirus from northern Australia suppresses replication of West Nile virus and Murray Valley encephalitis virus in co-infected mosquito cells.PLoS One. 2013;8(2):e56534. doi: 10.1371/journal.pone.0056534. Epub 2013 Feb 27. PLoS One. 2013. PMID: 23460804 Free PMC article.
-
Unleashing Nature's Allies: Comparing the Vertical Transmission Dynamics of Insect-Specific and Vertebrate-Infecting Flaviviruses in Mosquitoes.Viruses. 2024 Sep 23;16(9):1499. doi: 10.3390/v16091499. Viruses. 2024. PMID: 39339975 Free PMC article. Review.
-
Molecular evolution of the insect-specific flaviviruses.J Gen Virol. 2012 Feb;93(Pt 2):223-234. doi: 10.1099/vir.0.036525-0. Epub 2011 Oct 19. J Gen Virol. 2012. PMID: 22012464 Free PMC article. Review.
Cited by
-
Codon Usage and Context Analysis of Genes Modulated during SARS-CoV-2 Infection and Dental Inflammation.Vaccines (Basel). 2022 Nov 6;10(11):1874. doi: 10.3390/vaccines10111874. Vaccines (Basel). 2022. PMID: 36366382 Free PMC article.
-
In human astrocytes neurotropic flaviviruses increase autophagy, yet their replication is autophagy-independent.Cell Mol Life Sci. 2022 Oct 25;79(11):566. doi: 10.1007/s00018-022-04578-7. Cell Mol Life Sci. 2022. PMID: 36283999 Free PMC article.
-
Natural vertical transmission of dengue virus in Latin America and the Caribbean: highlighting its detection limitations and potential significance.Parasit Vectors. 2023 Nov 28;16(1):442. doi: 10.1186/s13071-023-06043-1. Parasit Vectors. 2023. PMID: 38017450 Free PMC article. Review.
-
Old Drugs with New Tricks: Efficacy of Fluoroquinolones to Suppress Replication of Flaviviruses.Viruses. 2020 Sep 13;12(9):1022. doi: 10.3390/v12091022. Viruses. 2020. PMID: 32933138 Free PMC article.
-
Vertical Transmission of Zika Virus in Aedes aegypti Mosquitoes.Am J Trop Med Hyg. 2016 Nov 2;95(5):1169-1173. doi: 10.4269/ajtmh.16-0448. Epub 2016 Aug 29. Am J Trop Med Hyg. 2016. PMID: 27573623 Free PMC article.
References
-
- Lindenbach B.D., Thiel H.-J., Rice C.M. Flaviviridae: The viruses and their replication. In: Knipe D.M., Howley P.M., editors. Fields Virology. 5th ed. Lippincott Williams and Wilkins; Philadelphia, PA, USA: 2007. pp. 1101–1152.
-
- Gubler D.J., Kuno G., Markoff L. Flaviviruses. In: Knipe D.M., Howley P.M., editors. Fields Virology. 5th ed. Lippincott Williams and Wilkins; Philadelphia, PA, USA: 2007. pp. 1153–1252.
-
- Johnson H.N. Ecological implications of antigenically related mammalian viruses for which arthropod vectors are unknown and avian associated soft tick viruses. Jpn. J. Med. Sci. Biol. 1967;20(Suppl):160–166. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
