

Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms
- PMID: 25866248
- PMCID: PMC4402149
- DOI: 10.1016/j.molcel.2015.03.005
Single mammalian cells compensate for differences in cellular volume and DNA copy number through independent global transcriptional mechanisms
Abstract
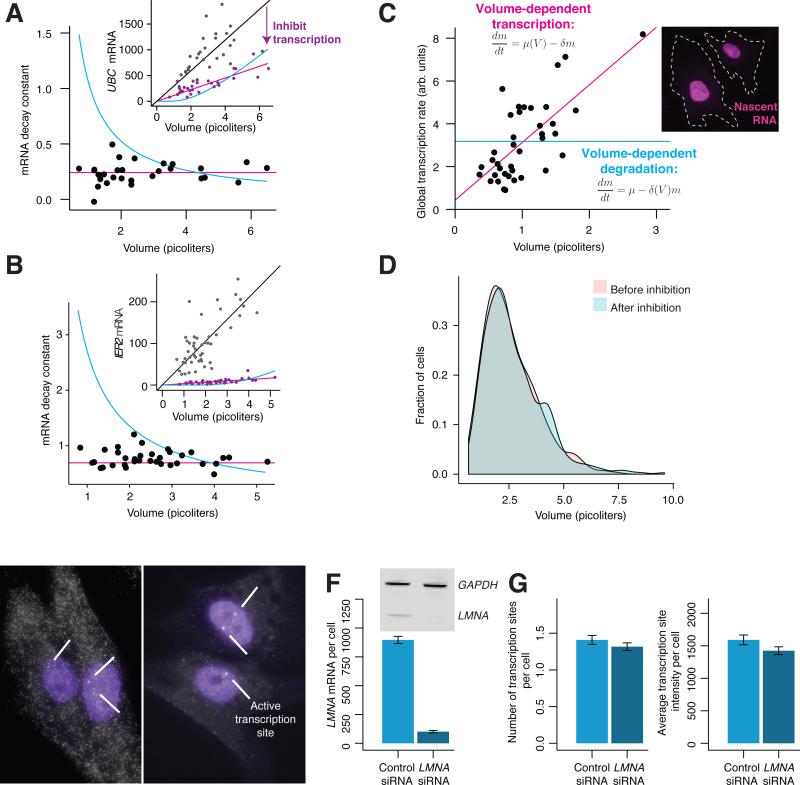
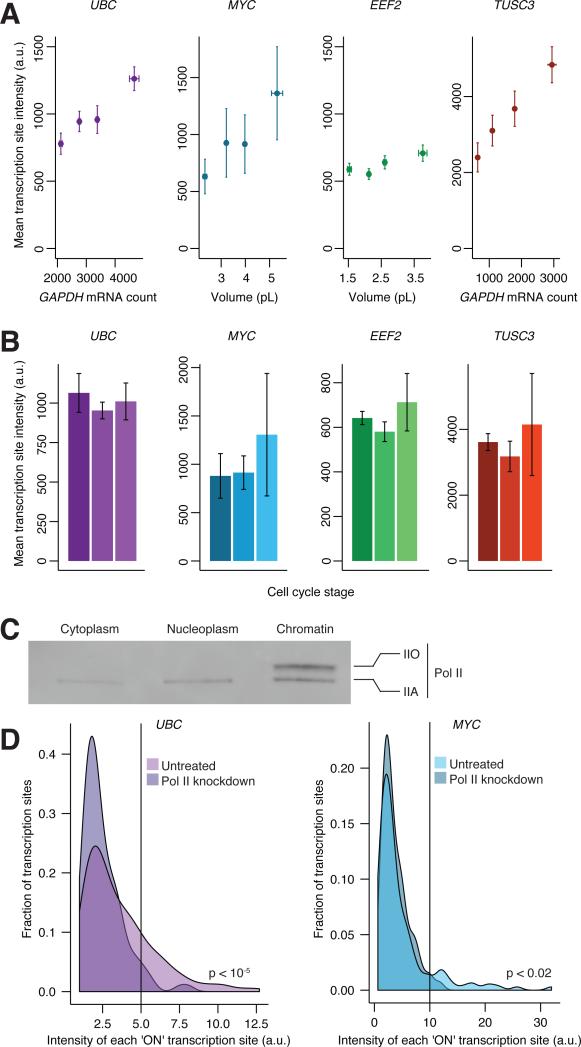
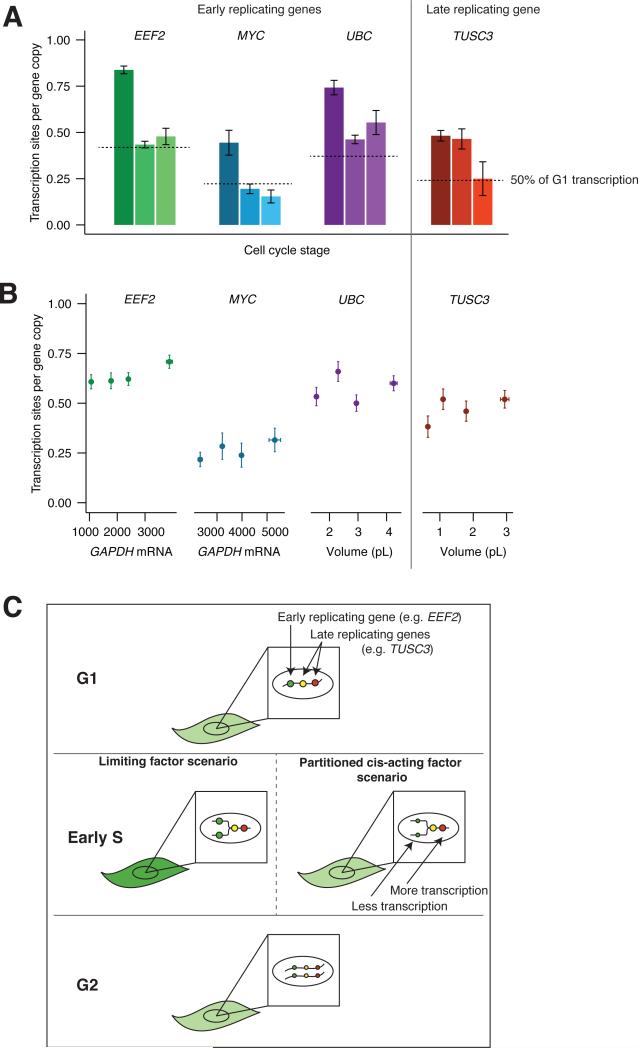
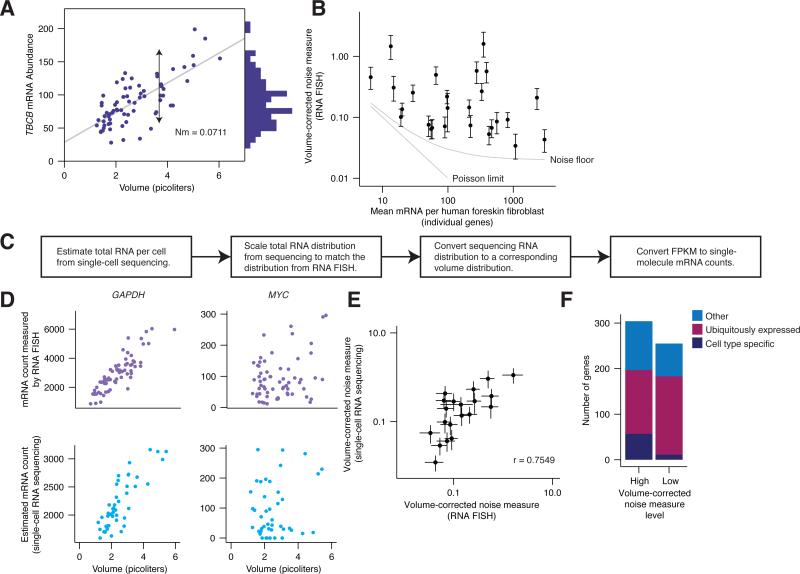
Individual mammalian cells exhibit large variability in cellular volume, even with the same absolute DNA content, and so must compensate for differences in DNA concentration in order to maintain constant concentration of gene expression products. Using single-molecule counting and computational image analysis, we show that transcript abundance correlates with cellular volume at the single-cell level due to increased global transcription in larger cells. Cell fusion experiments establish that increased cellular content itself can directly increase transcription. Quantitative analysis shows that this mechanism measures the ratio of cellular volume to DNA content, most likely through sequestration of a transcriptional factor to DNA. Analysis of transcriptional bursts reveals a separate mechanism for gene dosage compensation after DNA replication that enables proper transcriptional output during early and late S phase. Our results provide a framework for quantitatively understanding the relationships among DNA content, cell size, and gene expression variability in single cells.
Copyright © 2015 Elsevier Inc. All rights reserved.
Figures



Similar articles
-
Spatial organization of the somatosensory cortex revealed by osmFISH.Nat Methods. 2018 Nov;15(11):932-935. doi: 10.1038/s41592-018-0175-z. Epub 2018 Oct 30. Nat Methods. 2018. PMID: 30377364
-
Control of Transcript Variability in Single Mammalian Cells.Cell. 2015 Dec 17;163(7):1596-610. doi: 10.1016/j.cell.2015.11.018. Cell. 2015. PMID: 26687353
-
Dense transcript profiling in single cells by image correlation decoding.Nat Methods. 2016 Aug;13(8):657-60. doi: 10.1038/nmeth.3895. Epub 2016 Jun 6. Nat Methods. 2016. PMID: 27271198 Free PMC article.
-
Investigating transcriptional states at single-cell-resolution.Curr Opin Biotechnol. 2013 Feb;24(1):69-78. doi: 10.1016/j.copbio.2012.09.013. Epub 2012 Oct 17. Curr Opin Biotechnol. 2013. PMID: 23084076 Review.
-
MEMOIR: A Novel System for Neural Lineage Tracing.Neurosci Bull. 2017 Dec;33(6):763-765. doi: 10.1007/s12264-017-0161-y. Epub 2017 Aug 5. Neurosci Bull. 2017. PMID: 28780643 Free PMC article. Review. No abstract available.
Cited by
-
Cell size distribution of lineage data: analytic results and parameter inference.iScience. 2021 Feb 24;24(3):102220. doi: 10.1016/j.isci.2021.102220. eCollection 2021 Mar 19. iScience. 2021. PMID: 33748708 Free PMC article.
-
Coupling gene expression dynamics to cell size dynamics and cell cycle events: Exact and approximate solutions of the extended telegraph model.iScience. 2022 Dec 7;26(1):105746. doi: 10.1016/j.isci.2022.105746. eCollection 2023 Jan 20. iScience. 2022. PMID: 36619980 Free PMC article.
-
A Spatiotemporal DNA Endoploidy Map of the Arabidopsis Root Reveals Roles for the Endocycle in Root Development and Stress Adaptation.Plant Cell. 2018 Oct;30(10):2330-2351. doi: 10.1105/tpc.17.00983. Epub 2018 Aug 16. Plant Cell. 2018. PMID: 30115738 Free PMC article.
-
Role of Chromatin Replication in Transcriptional Plasticity, Cell Differentiation and Disease.Genes (Basel). 2022 Jun 2;13(6):1002. doi: 10.3390/genes13061002. Genes (Basel). 2022. PMID: 35741764 Free PMC article. Review.
-
Periodic synchronization of isolated network elements facilitates simulating and inferring gene regulatory networks including stochastic molecular kinetics.BMC Bioinformatics. 2022 Jan 5;23(1):13. doi: 10.1186/s12859-021-04541-6. BMC Bioinformatics. 2022. PMID: 34986805 Free PMC article.
References
-
- Bar-Even A, Paulsson J, Maheshri N, Carmi M, O'Shea E, Pilpel Y, Barkai N. Noise in protein expression scales with natural protein abundance. Nat. Genet. 2006;38:636–643. - PubMed
-
- Brennecke P, Anders S, Kim JK, Kołodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nature Methods. 2013;10:1093–1095. - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
