

Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension
- PMID: 25825391
- PMCID: PMC4459813
- DOI: 10.15252/emmm.201404511
Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension
Abstract
Iron-sulfur (Fe-S) clusters are essential for mitochondrial metabolism, but their regulation in pulmonary hypertension (PH) remains enigmatic. We demonstrate that alterations of the miR-210-ISCU1/2 axis cause Fe-S deficiencies in vivo and promote PH. In pulmonary vascular cells and particularly endothelium, hypoxic induction of miR-210 and repression of the miR-210 targets ISCU1/2 down-regulated Fe-S levels. In mouse and human vascular and endothelial tissue affected by PH, miR-210 was elevated accompanied by decreased ISCU1/2 and Fe-S integrity. In mice, miR-210 repressed ISCU1/2 and promoted PH. Mice deficient in miR-210, via genetic/pharmacologic means or via an endothelial-specific manner, displayed increased ISCU1/2 and were resistant to Fe-S-dependent pathophenotypes and PH. Similar to hypoxia or miR-210 overexpression, ISCU1/2 knockdown also promoted PH. Finally, cardiopulmonary exercise testing of a woman with homozygous ISCU mutations revealed exercise-induced pulmonary vascular dysfunction. Thus, driven by acquired (hypoxia) or genetic causes, the miR-210-ISCU1/2 regulatory axis is a pathogenic lynchpin causing Fe-S deficiency and PH. These findings carry broad translational implications for defining the metabolic origins of PH and potentially other metabolic diseases sharing similar underpinnings.
Keywords: endothelial; iron–sulfur; metabolism; microRNA; mitochondria.
© 2015 The Authors. Published under the terms of the CC BY 4.0 license.
Figures
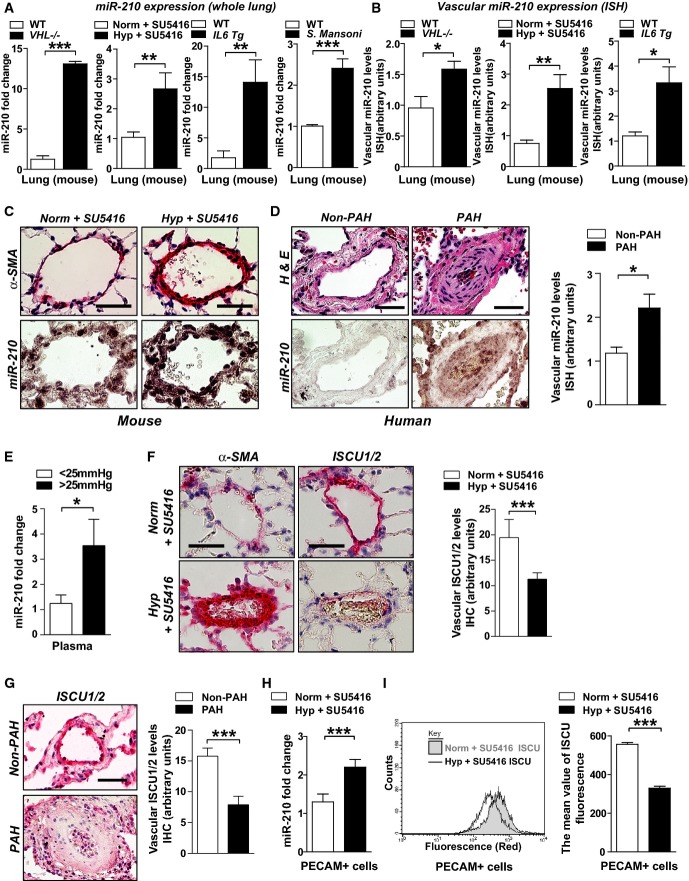
A RT–qPCR reveals that miR-210 was increased in lungs of mice with PH triggered by various conditions: VHL−/− as compared with VHL+/+ mice (N = 4/group), ***P < 0.0001 (first graph); hypoxia + SU5416 (Hyp + SU5416) (N = 6/group) as compared with normoxia + SU5416 (Norm + SU5416) (N = 7/group), **P = 0.0015 (second graph); Il6 transgenic versus littermate control mice (N = 4/group), **P = 0.0097 (third graph); and S. mansoni-infected mice (N = 4) compared with non-infected control mice (N = 5), ***P < 0.0001 (fourth graph).
B From animal subjects in (A), in situ hybridization (ISH, purple stain) revealed increased miR-210 in < 100-μm pulmonary vessels of mice suffering from PH, *P = 0.0493 for first graph, **P = 0.0015 for second graph, *P = 0.0391 for third graph.
C Representative ISH stain of miR-210 in < 100 μm pulmonary vessels of mice (bottom micrographs) exposed to Hyp + SU5416 compared with Norm + SU5416 (α-smooth muscle actin stain from serial sections, top row of micrographs).
D Increased miR-210 in < 200-μm remodeled pulmonary vessels of patients suffering from PAH (N = 19, Supplementary Table S1) as compared with non-PAH donor control lung (N = 10). Serial staining with hematoxylin and eosin is displayed in the top row of micrographs; quantification of miRNA ISH, right graph, *P = 0.0167.
E Increased levels of miR-210 in plasma drawn from the pulmonary circulation (pulmonary capillary wedge position, PCWP) of patients with elevated mean pulmonary arterial pressures (mean PAP ≥ 25 mmHg) compared with control subjects (mPAP < 25 mmHg, N = 5/group, demographics in Supplementary Table S2), *P = 0.0357.
F, G From animals in (C) and humans in (D), immunohistochemistry (IHC) revealed that the miR-210 targets ISCU1/2 were reciprocally down-regulated in miR-210-enriched remodeled pulmonary vessels—namely in PH mice exposed to Hyp + SU5416 (F, ***P = 0.0002) and in human PAH patients (G, ***P = 0.0008).
H, I miR-210 expression (H) was increased (***P = 0.0003), and ISCU1/2 expression (I) was decreased (***P < 0.0001) in PECAM+ pulmonary vascular endothelial cells isolated from PH mice (Hyp + SU5416) as compared with control (Norm + SU5416) (N = 4/group, left bars).
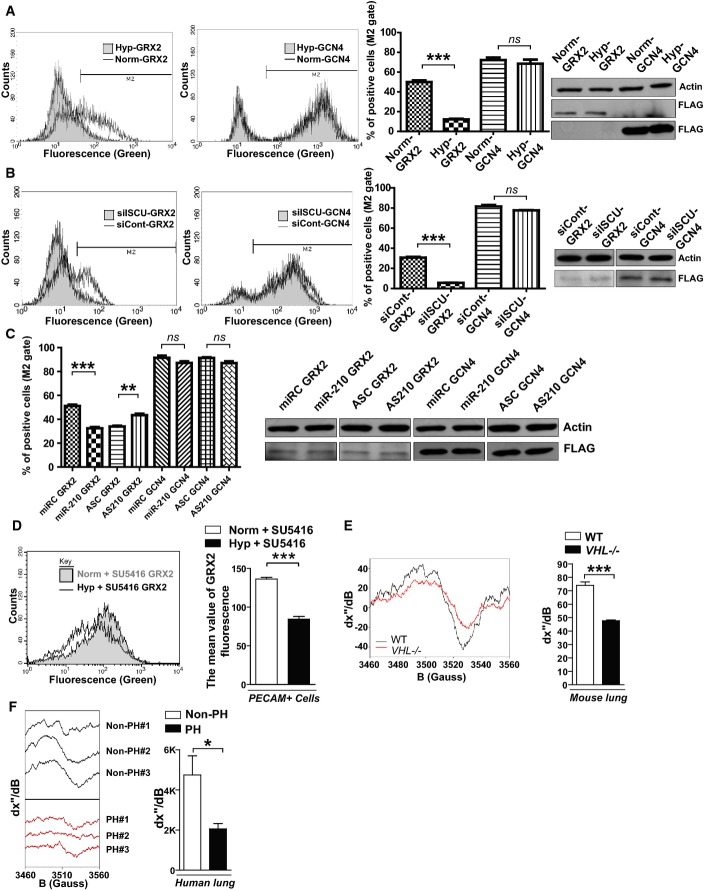
After lentiviral delivery of GCN4 or GRX2 sensor genes to human PAECs, cellular fluorescence was measured by flow cytometry. Unlike control GCN4 sensors that homodimerized independent of Fe-S levels and induced consistent fluorescence, fluorescence derived from the GRX2 Fe-S-dependent sensors decreased in hypoxia, as displayed in representative flow cytometric plots (left) and by quantification of percentage of positive cells [(cell number in M2 gate)/(total cell number) × 100]. Immunoblotting revealed consistent expression of either GRX2 or GCN4 sensors (FLAG-tagged) in hypoxia (Hyp) compared with normoxia (Norm) (N = 3, ***P < 0.0001 for GRX2; N = 3, NS P = 0.1848 for GCN4).
In contrast to consistent fluorescence from control GCN4 sensors, Fe-S-dependent GRX2 sensor fluorescence was decreased by siRNA knockdown of ISCU1/2 (siISCU) as compared with control (siCont) (N = 3, ***P < 0.0001 for GRX2; N = 3, NS P = 0.1790 for GCN4).
GRX2, but not GCN4, sensor fluorescence was decreased after transfection of miR-210 oligonucleotide mimic (miR-210) as compared with control (miRC) (N = 3, ***P = 0.0002 for GRX2; N = 3, NS P = 0.0913 for GCN4). During hypoxic exposure, GRX2, but not GCN4, sensor fluorescence was increased after transfection of an antisense miR-210 inhibitor (AS210) as compared with control (ASC) (N = 3, **P = 0.003 for GRX2; N = 3, NS P = 0.1194 for GCN4).
After lentiviral delivery of sensor genes, Fe-S-dependent GRX2 sensor fluorescence was decreased in PECAM-positive cells from PH mouse lung (Hyp + SU5416) as compared with control non-PH mouse lung (Norm + SU5416, N = 3, ***P < 0.001).
By electron paramagnetic resonance (EPR) spectroscopy (representative VHL−/− versus control lung), Fe-S cluster signal was decreased (left graph) in VHL−/− mice lung tissue (VHL−/−, N = 3) as compared with WT control lung tissues (WT, N = 5), ***P = 0.0003.
By EPR, Fe-S-specific signal was decreased (right graph) in human PH-diseased lung tissue harvested at lung transplantation (PH, N = 3, Supplementary Table S3) compared with non-PH control donor lung tissues (control, N = 3), *P = 0.048.
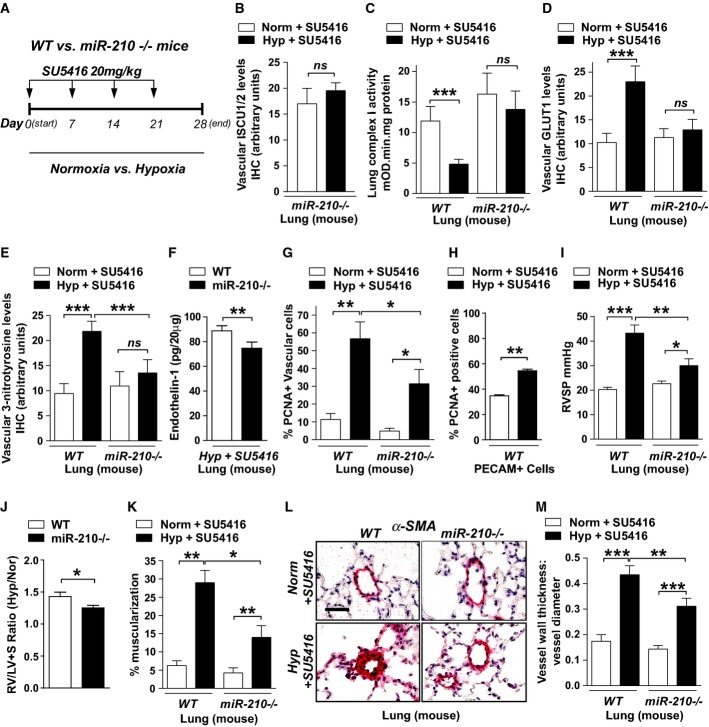
A Schema of comparing miR-210−/− and WT mice after hypoxia + SU5416 (PH) versus normoxia + SU5416 (control).
B By immunohistochemistry (IHC), ISCU1/2 was unchanged in < 100 μm pulmonary vessels of miR-210−/− mice exposed to Hyp + SU5416 (N = 6) versus Norm + SU5416 (N = 7), NS P = 0.2833.
C Fe-S-dependent Complex I-specific activity was decreased in WT PH mice as compared with control (N = 7/group, left bars, ***P < 0.0001), but activity was preserved in the lungs of miR-210−/− mice in either condition (N = 8/group, right bars), NS P = 0.3693.
D IHC demonstrated that pulmonary vascular GLUT1 was increased in WT PH mice (left bars, ***P = 0.0006), but GLUT1 was unchanged in miR-210−/− mice in either condition (right bars, NS P = 0.9967).
E 3-nitrotyrosine (3-NT) was increased in pulmonary vessels of WT PH mice but was reduced in miR-210−/− mice in either condition (N = 6/group) (***P < 0.0001, NS P = 0.4087).
F In PH conditions, endothelin-1 was decreased in miR-210−/− lung tissue compared with WT tissue (N = 5/group), **P = 0.007.
G PCNA was increased in WT PH pulmonary vessels but was decreased in miR-210−/− tissue exposed to either condition (N = 5/group) (**P = 0.0096, *P = 0.0124, *P = 0.0263 for miR-210−/−).
H PCNA was increased in PECAM+ endothelial cells from PH versus non-PH mice (N = 3/group, **P = 0.0052).
I Unlike WT mice (black bars) demonstrating increased right ventricular systolic pressure (RVSP) after Hyp + SU5416 (N = 10) versus Norm + SU5416 (N = 11), hemodynamic dysregulation was significantly alleviated in miR-210−/− mice (white bars, N = 11) (***P < 0.0001, *P = 0.0258, **P = 0.0059).
J Compared with WT controls (N = 9), miR-210−/− mice (N = 8) displayed a blunted increase of the Fulton index (RV/LV + S) under PH versus baseline conditions (expressed as a ratio of RV/LV + S under Hyp + SU5416 versus Norm + SU5416, *P = 0.031).
K–M Under PH (black bars, N = 8/group) versus baseline conditions (white bars, N = 6/group), pulmonary vascular remodeling was alleviated in miR-210−/− mice, as visualized via histology (L), and confirmed by decreased % arteriolar muscularization (K, **P = 0.001, **P = 0.0045 for miR-210−/−, *P = 0.0158) and decreased vessel wall thickness (M, ***P < 0.0001, **P = 0.0086).
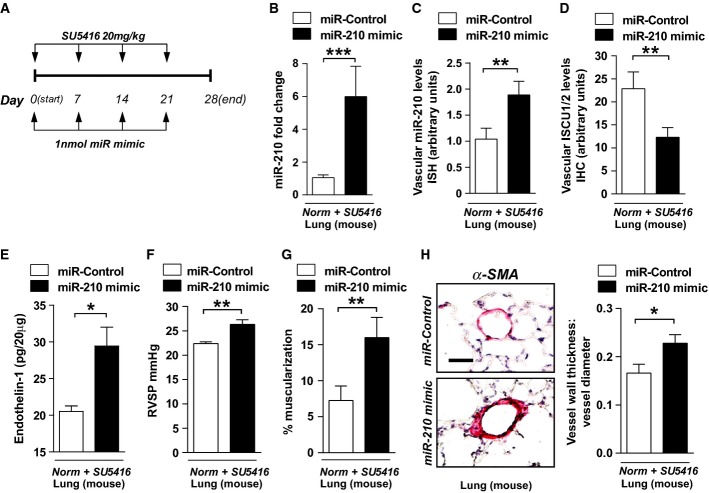
A Schema for forced miR-210 expression in vivo (N = 8/group).
B, C Intrapharyngeal delivery of miR-210 mimic increased miR-210 in whole lung (***P = 0.0002) (B) and in < 100-μm pulmonary vessels (**P = 0.007) (C).
D miR-210 mimic also repressed ISCU1/2 levels in those same caliber vessels (N = 6/group), **P = 0.0022.
E Endothelin-1 was increased in mouse lung tissue after delivery of miR-210 mimic (N = 6/group), *P = 0.0368.
F–H miR-210 mimic delivery increased RVSP (N = 8/group) (F) and vascular remodeling (α-smooth muscle actin stain, α-SMA), as evidenced by increased percent of muscularized (< 100 μm) pulmonary vessels (G) and increased medial thickening relative to vessel diameter when compared with miR-Control (H) (N = 6/group). **P = 0.0021 for (F), **P = 0.0072 for (G), *P = 0.0335 for (H).
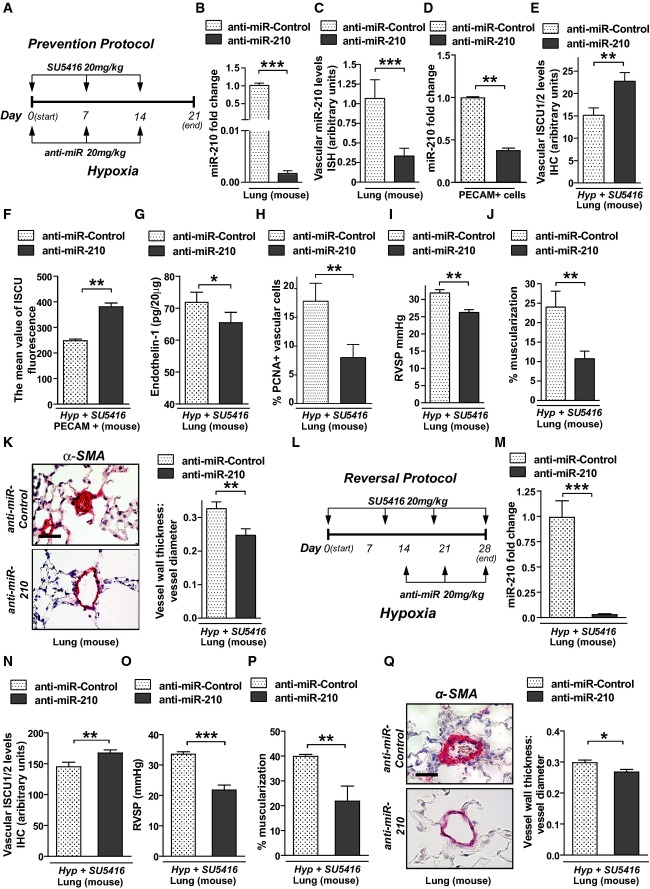
A Schema of the strategy to determine whether antisense inhibition of miR-210 (anti-miR-210) prevents PH secondary to Hyp + SU5416 exposure (“prevention protocol”).
B–D Intravenous delivery of anti-miR-210 down-regulated miR-210 expression in whole lung tissue (N = 7/group), ***P < 0.0001 (B), the pulmonary vasculature as assessed by in situ staining (N = 7/group), ***P = 0.0003 (C), and PECAM+ vascular endothelial cells derived from whole lung tissue (N = 4/group), **P = 0.0013 (D).
E Correspondingly, anti-miR-210 delivery resulted in preservation of ISCU1/2 expression in < 100-μm pulmonary vessels as compared to anti-miR-Control (N = 5/group), **P = 0.0011.
F Similarly, ISCU1/2 expression was preserved in PECAM+ endothelial cells derived from mice treated with anti-miR-210 (N = 4/group), **P = 0.0016.
G Endothelin-1 was decreased in lung tissue from mice treated with anti-miR-210 during Hyp + SU5416 exposure (N = 6/group), *P = 0.0181.
H As assessed by in situ immunofluorescence, PCNA was decreased in pulmonary vessels (< 100 μm) after anti-miR-210 delivery, **P = 0.001.
I–K Anti-miR-210 delivery (N = 10/group) ameliorated the elevation of RVSP, **P = 0.0031 (i) and vascular remodeling, as reflected by increased percent of muscularized arterioles (N = 8/group, **P = 0.0091) (J) and increased medial thickness relative to vessel diameter in < 100-μm pulmonary vessels (N = 6/group, **P = 0.0021) (K).
L Schema of the strategy to determine whether anti-miR-210 improves existing PH (“reversal protocol”).
M, N Pharmacologic inhibition of miR-210 (N = 7/group) down-regulated pulmonary expression of miR-210, ***P < 0.0001 (M), and preserved ISCU1/2 expression in < 100-μm pulmonary vessels, **P = 0.0015 (N).
O–Q In contrast to anti-miR-Control (N = 8), anti-miR-210 ameliorated the elevation of RVSP (N = 10/group, ***P < 0.0001) (O) and pulmonary vascular remodeling (N = 7/group, **P = 0.0022, *P = 0.0167) (P–Q).
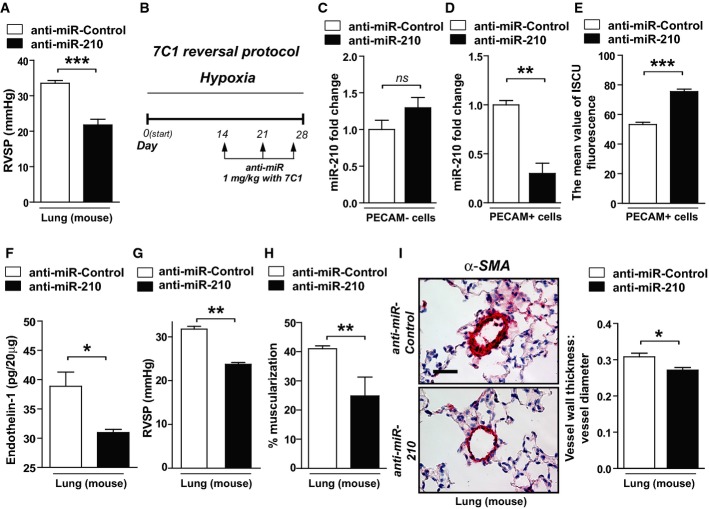
A Following the schema of Fig5A in the presence of hypoxia but the absence of SU5416, intravenous delivery of anti-miR-210 ameliorated the elevation of RVSP compared with anti-miR-Control (N = 6/group, ***P = 0.001).
B Schema of the strategy to determine whether delivery of anti-miR-210 specifically to the vascular endothelium via packaging with 7C1 nanoparticles improves hypoxia-induced PH (“7C1 reversal protocol”).
C, D Consistent with specific delivery to the vascular endothelium after intravenous administration (Dahlman et al, 2014), 7C1-mediated delivery of anti-miR-210 (N = 6/group) decreased miR-210 expression in PECAM+ pulmonary vascular endothelial cells, **P = 0.0013, (D) but not PECAM-negative pulmonary cells, NS P = 0.3775 (C).
E Correspondingly, endothelial-specific anti-miR-210 (N = 6/group) increased ISCU1/2 expression in PECAM+ cells even in the presence of chronic hypoxia, ***P = 0.0003.
F–I Delivery of anti-miR-210 (N = 5/group) decreased endothelin-1 in hypoxic mouse lung tissue, *P = 0.0198 (F). As a result, in contrast to anti-miR-Control (N = 8), endothelial-specific anti-miR-210 ameliorated the elevation of RVSP (N = 6), **P = 0.0024 (G), and pulmonary vascular remodeling (N = 6/group), **P = 0.0021, *P = 0.0117 (H–I).
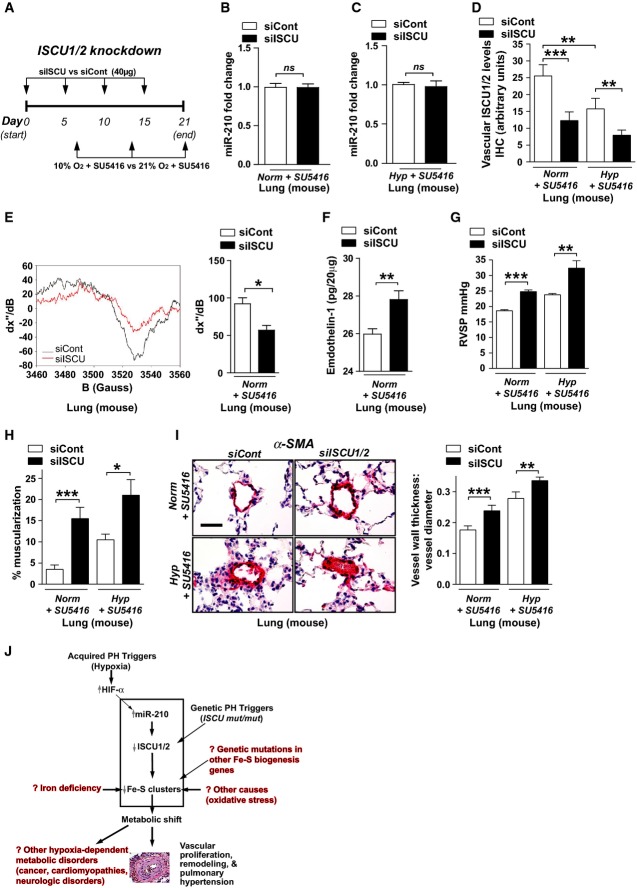
A Schema of the strategy for pharmacologic inhibition of siRNA knockdown of ISCU1/2 in the pulmonary vasculature of mice in vivo by Staramine-mPEG-mediated intravenous delivery.
B, C Pharmacologic inhibition of ISCU (N = 8/group) did not influence miR-210 expression in the lungs either after Norm + SU5415, NS P = 0.8634 (B) or Hyp + SU5416, NS P = 0.9786 (C).
D As compared with control siRNA (siCont) where ISCU1/2 expression was decreased after exposure to Hyp + SU5416 (white bars), pulmonary vascular delivery of siRNA specific for ISCU1/2 (siISCU) via Staramine-mPEG nanocomplexes (Polach et al, 2012) down-regulated ISCU1/2 in < 100-μm pulmonary vessels in Norm + SU5416 and Hyp + SU5416 (N = 5/group, ***P = 0.0006, **P = 0.0019 for Hyp + SU5416, **P = 0.0029 for siCont).
E By electron paramagnetic resonance (EPR) spectroscopy (representative siCont versus siISCU lung), Fe-S cluster signal was decreased (quantitative graph, right) in siISCU mouse lung tissue (N = 4 mice) compared with siCont (N = 5 mice), *P = 0.0143.
F Endothelin-1 was increased in lung tissue after siISCU delivery under normoxic conditions (N = 5/group), **P = 0.0027.
G–I In the absence (left bars) and presence (right bars) of Hyp + SU5416 for 2 weeks, siISCU (black bars) induced elevations in RVSP (***P < 0.0001, **P = 0.0031) (G) and increased pulmonary vascular remodeling in < 100-μm pulmonary vessels (H, I) as compared with siCont (N = 5/group, ***P = 0.0003, *P = 0.0114 for H; ***P = 0.0006, **P = 0.0098 for I).
J Model of acquired and genetic Fe-S deficiency as a central cause of metabolic dysfunction and PH in mice and humans. Among others, potential translational implications are highlighted in red text.
Comment in
-
New insights into the pathology of pulmonary hypertension: implication of the miR-210/ISCU1/2/Fe-S axis.EMBO Mol Med. 2015 Jun;7(6):689-91. doi: 10.15252/emmm.201505160. EMBO Mol Med. 2015. PMID: 25851536 Free PMC article.
Similar articles
-
New insights into the pathology of pulmonary hypertension: implication of the miR-210/ISCU1/2/Fe-S axis.EMBO Mol Med. 2015 Jun;7(6):689-91. doi: 10.15252/emmm.201505160. EMBO Mol Med. 2015. PMID: 25851536 Free PMC article.
-
MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2.Cell Metab. 2009 Oct;10(4):273-84. doi: 10.1016/j.cmet.2009.08.015. Cell Metab. 2009. PMID: 19808020 Free PMC article.
-
MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells.Cell Death Dis. 2014 Feb 27;5(2):e1090. doi: 10.1038/cddis.2014.60. Cell Death Dis. 2014. PMID: 24577088 Free PMC article.
-
Mitochondrial iron-sulfur protein biogenesis and human disease.Biochimie. 2014 May;100:61-77. doi: 10.1016/j.biochi.2014.01.010. Epub 2014 Jan 23. Biochimie. 2014. PMID: 24462711 Review.
-
Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease.Biochemistry. 2010 Jun 22;49(24):4945-56. doi: 10.1021/bi1004798. Biochemistry. 2010. PMID: 20481466 Free PMC article. Review.
Cited by
-
Therapeutic RNA Delivery for COVID and Other Diseases.Adv Healthc Mater. 2021 Aug;10(15):e2002022. doi: 10.1002/adhm.202002022. Epub 2021 Mar 4. Adv Healthc Mater. 2021. PMID: 33661555 Free PMC article. Review.
-
Metabolic dysfunction in pulmonary hypertension: from basic science to clinical practice.Eur Respir Rev. 2017 Dec 20;26(146):170094. doi: 10.1183/16000617.0094-2017. Print 2017 Dec 31. Eur Respir Rev. 2017. PMID: 29263174 Free PMC article. Review.
-
Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension.J Clin Invest. 2016 Sep 1;126(9):3313-35. doi: 10.1172/JCI86387. Epub 2016 Aug 22. J Clin Invest. 2016. PMID: 27548520 Free PMC article.
-
Rats with a Human Mutation of NFU1 Develop Pulmonary Hypertension.Am J Respir Cell Mol Biol. 2020 Feb;62(2):231-242. doi: 10.1165/rcmb.2019-0065OC. Am J Respir Cell Mol Biol. 2020. PMID: 31461310 Free PMC article.
-
Pulmonary Arterial Hypertension: Iron Matters.Front Physiol. 2018 May 31;9:641. doi: 10.3389/fphys.2018.00641. eCollection 2018. Front Physiol. 2018. PMID: 29904352 Free PMC article. Review.
References
-
- Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, McGoon M, Naeije R, Olschewski H, Oudiz RJ, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S55–S66. - PubMed
-
- Barclay AR, Sholler G, Christodolou J, Shun A, Arbuckle S, Dorney S, Stormon MO. Pulmonary hypertension–a new manifestation of mitochondrial disease. J Inherit Metab Dis. 2005;28:1081–1089. - PubMed
-
- Beinert H, Holm RH, Munck E. Iron–sulfur clusters: nature's modular, multipurpose structures. Science. 1997;277:653–659. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 HL096834/HL/NHLBI NIH HHS/United States
- R01 HL122596/HL/NHLBI NIH HHS/United States
- 098051/WT_/Wellcome Trust/United Kingdom
- R37 EB000244/EB/NIBIB NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 DE016516/DE/NIDCR NIH HHS/United States
- R01-DE016516-06/DE/NIDCR NIH HHS/United States
- R01 EB000244/EB/NIBIB NIH HHS/United States
- U54 CA151884/CA/NCI NIH HHS/United States
- U54-CA151884/CA/NCI NIH HHS/United States
- ImNIH/Intramural NIH HHS/United States
- K08 HL105536/HL/NHLBI NIH HHS/United States
- K08HL096834/HL/NHLBI NIH HHS/United States
- EB000244/EB/NIBIB NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
