

Mitophagy and cancer
- PMID: 25810907
- PMCID: PMC4373087
- DOI: 10.1186/s40170-015-0130-8
Mitophagy and cancer
Abstract
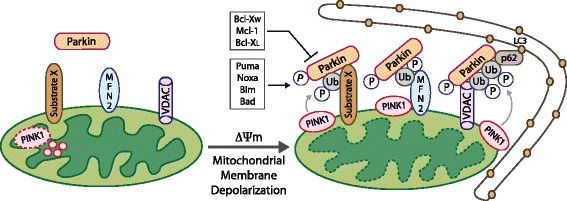
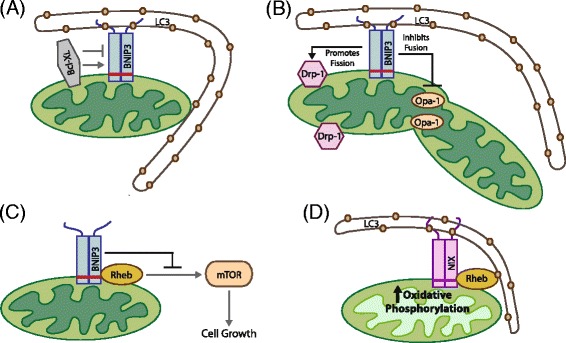
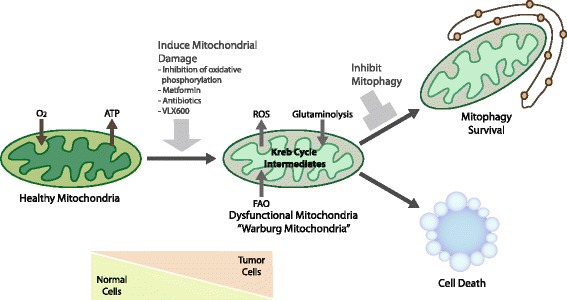
Mitophagy is a selective form of macro-autophagy in which mitochondria are selectively targeted for degradation in autophagolysosomes. Mitophagy can have the beneficial effect of eliminating old and/or damaged mitochondria, thus maintaining the integrity of the mitochondrial pool. However, mitophagy is not only limited to the turnover of dysfunctional mitochondria but also promotes reduction of overall mitochondrial mass in response to certain stresses, such as hypoxia and nutrient starvation. This prevents generation of reactive oxygen species and conserves valuable nutrients (such as oxygen) from being consumed inefficiently, thereby promoting cellular survival under conditions of energetic stress. The failure to properly modulate mitochondrial turnover in response to oncogenic stresses has been implicated both positively and negatively in tumorigenesis, while the potential of targeting mitophagy specifically as opposed to autophagy in general as a therapeutic strategy remains to be explored. The challenges and opportunities that come with our heightened understanding of the role of mitophagy in cancer are reviewed here.
Keywords: Autophagosomes; BNIP3; Mitochondrial dysfunction; Mitophagy; NIX; Parkin.
Figures
Similar articles
-
Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys.Autophagy. 2019 Dec;15(12):2142-2162. doi: 10.1080/15548627.2019.1615822. Epub 2019 May 22. Autophagy. 2019. PMID: 31066324 Free PMC article.
-
Mitophagy and oral cancers.Natl J Maxillofac Surg. 2022 Jan-Apr;13(1):11-19. doi: 10.4103/njms.NJMS_123_20. Epub 2022 Apr 20. Natl J Maxillofac Surg. 2022. PMID: 35911821 Free PMC article. Review.
-
Autophagy capacity and sub-mitochondrial heterogeneity shape Bnip3-induced mitophagy regulation of apoptosis.Cell Commun Signal. 2015 Aug 8;13:37. doi: 10.1186/s12964-015-0115-9. Cell Commun Signal. 2015. PMID: 26253153 Free PMC article.
-
Mitophagy: Link to cancer development and therapy.Biochem Biophys Res Commun. 2017 Jan 15;482(3):432-439. doi: 10.1016/j.bbrc.2016.10.088. Epub 2017 Feb 3. Biochem Biophys Res Commun. 2017. PMID: 28212727 Review.
-
Mitochondria autophagy is induced after hypoxic/ischemic stress in a Drp1 dependent manner: the role of inhibition of Drp1 in ischemic brain damage.Neuropharmacology. 2014 Nov;86:103-15. doi: 10.1016/j.neuropharm.2014.07.002. Epub 2014 Jul 10. Neuropharmacology. 2014. PMID: 25018043
Cited by
-
Mitophagy in carcinogenesis and cancer treatment.Discov Oncol. 2021 Dec 1;12(1):58. doi: 10.1007/s12672-021-00454-1. Discov Oncol. 2021. PMID: 35201480 Free PMC article. Review.
-
Is There a Mitochondrial Protection via Remote Ischemic Conditioning in Settings of Anticancer Therapy Cardiotoxicity?Curr Heart Fail Rep. 2024 Aug;21(4):292-304. doi: 10.1007/s11897-024-00658-w. Epub 2024 Mar 21. Curr Heart Fail Rep. 2024. PMID: 38512567 Free PMC article. Review.
-
Fanconi Anemia Proteins Function in Mitophagy and Immunity.Cell. 2016 May 5;165(4):867-81. doi: 10.1016/j.cell.2016.04.006. Epub 2016 Apr 28. Cell. 2016. PMID: 27133164 Free PMC article.
-
An Experimentally Induced Mutation in the UBA Domain of p62 Changes the Sensitivity of Cisplatin by Up-Regulating HK2 Localisation on the Mitochondria and Increasing Mitophagy in A2780 Ovarian Cancer Cells.Int J Mol Sci. 2021 Apr 13;22(8):3983. doi: 10.3390/ijms22083983. Int J Mol Sci. 2021. PMID: 33924293 Free PMC article.
-
Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis.EMBO Rep. 2015 Sep;16(9):1145-63. doi: 10.15252/embr.201540759. Epub 2015 Jul 31. EMBO Rep. 2015. PMID: 26232272 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
