

hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1
- PMID: 25799984
- PMCID: PMC4376666
- DOI: 10.1038/nature14280
hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1
Abstract
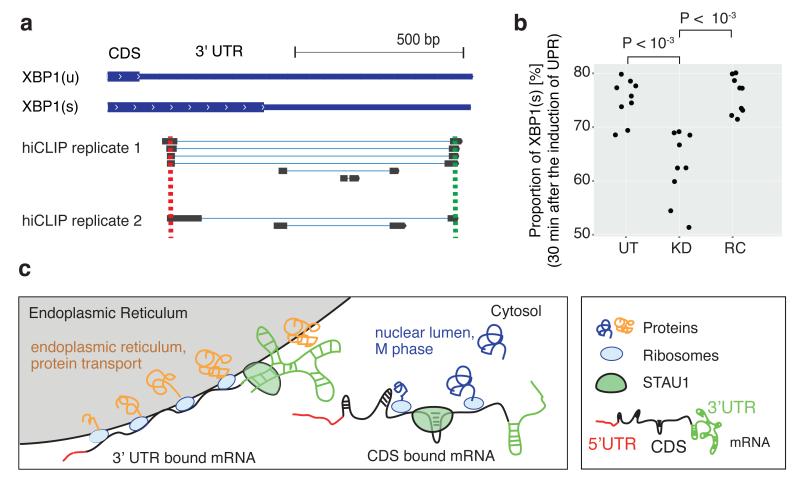
The structure of messenger RNA is important for post-transcriptional regulation, mainly because it affects binding of trans-acting factors. However, little is known about the in vivo structure of full-length mRNAs. Here we present hiCLIP, a biochemical technique for transcriptome-wide identification of RNA secondary structures interacting with RNA-binding proteins (RBPs). Using this technique to investigate RNA structures bound by Staufen 1 (STAU1) in human cells, we uncover a dominance of intra-molecular RNA duplexes, a depletion of duplexes from coding regions of highly translated mRNAs, an unexpected prevalence of long-range duplexes in 3' untranslated regions (UTRs), and a decreased incidence of single nucleotide polymorphisms in duplex-forming regions. We also discover a duplex spanning 858 nucleotides in the 3' UTR of the X-box binding protein 1 (XBP1) mRNA that regulates its cytoplasmic splicing and stability. Our study reveals the fundamental role of mRNA secondary structures in gene expression and introduces hiCLIP as a widely applicable method for discovering new, especially long-range, RNA duplexes.
Figures













Comment in
-
RNA. Detailed probing of RNA structure in vivo.Nat Rev Genet. 2015 May;16(5):255. doi: 10.1038/nrg3939. Epub 2015 Apr 9. Nat Rev Genet. 2015. PMID: 25854184 No abstract available.
Similar articles
-
CLIPing Staufen to secondary RNA structures: size and location matter!Bioessays. 2015 Oct;37(10):1062-6. doi: 10.1002/bies.201500052. Epub 2015 Aug 7. Bioessays. 2015. PMID: 26252431
-
lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3' UTRs via Alu elements.Nature. 2011 Feb 10;470(7333):284-8. doi: 10.1038/nature09701. Nature. 2011. PMID: 21307942 Free PMC article.
-
A computationally-enhanced hiCLIP atlas reveals Staufen1-RNA binding features and links 3' UTR structure to RNA metabolism.Nucleic Acids Res. 2023 May 8;51(8):3573-3589. doi: 10.1093/nar/gkad221. Nucleic Acids Res. 2023. PMID: 37013995 Free PMC article.
-
The role of secondary structures in the functioning of 3' untranslated regions of mRNA: A review of functions of 3' UTRs' secondary structures and hypothetical involvement of secondary structures in cytoplasmic polyadenylation in Drosophila.Bioessays. 2024 Mar;46(3):e2300099. doi: 10.1002/bies.202300099. Epub 2023 Dec 31. Bioessays. 2024. PMID: 38161240 Review.
-
Cataloguing and Selection of mRNAs Localized to Dendrites in Neurons and Regulated by RNA-Binding Proteins in RNA Granules.Biomolecules. 2020 Jan 22;10(2):167. doi: 10.3390/biom10020167. Biomolecules. 2020. PMID: 31978946 Free PMC article. Review.
Cited by
-
The expanding universe of ribonucleoproteins: of novel RNA-binding proteins and unconventional interactions.Pflugers Arch. 2016 Jun;468(6):1029-40. doi: 10.1007/s00424-016-1819-4. Epub 2016 May 10. Pflugers Arch. 2016. PMID: 27165283 Free PMC article. Review.
-
CircRNA-Protein Interactions in Muscle Development and Diseases.Int J Mol Sci. 2021 Mar 23;22(6):3262. doi: 10.3390/ijms22063262. Int J Mol Sci. 2021. PMID: 33806945 Free PMC article. Review.
-
Classification and clustering of RNA crosslink-ligation data reveal complex structures and homodimers.Genome Res. 2022 May;32(5):968-985. doi: 10.1101/gr.275979.121. Epub 2022 Mar 24. Genome Res. 2022. PMID: 35332099 Free PMC article.
-
Chemical RNA Cross-Linking: Mechanisms, Computational Analysis, and Biological Applications.JACS Au. 2023 Jan 16;3(2):316-332. doi: 10.1021/jacsau.2c00625. eCollection 2023 Feb 27. JACS Au. 2023. PMID: 36873678 Free PMC article. Review.
-
Gain of Additional BIRC3 Protein Functions through 3'-UTR-Mediated Protein Complex Formation.Mol Cell. 2019 May 16;74(4):701-712.e9. doi: 10.1016/j.molcel.2019.03.006. Epub 2019 Apr 1. Mol Cell. 2019. PMID: 30948266 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
