

Trametinib with or without vemurafenib in BRAF mutated non-small cell lung cancer
- PMID: 25706985
- PMCID: PMC4338247
- DOI: 10.1371/journal.pone.0118210
Trametinib with or without vemurafenib in BRAF mutated non-small cell lung cancer
Abstract
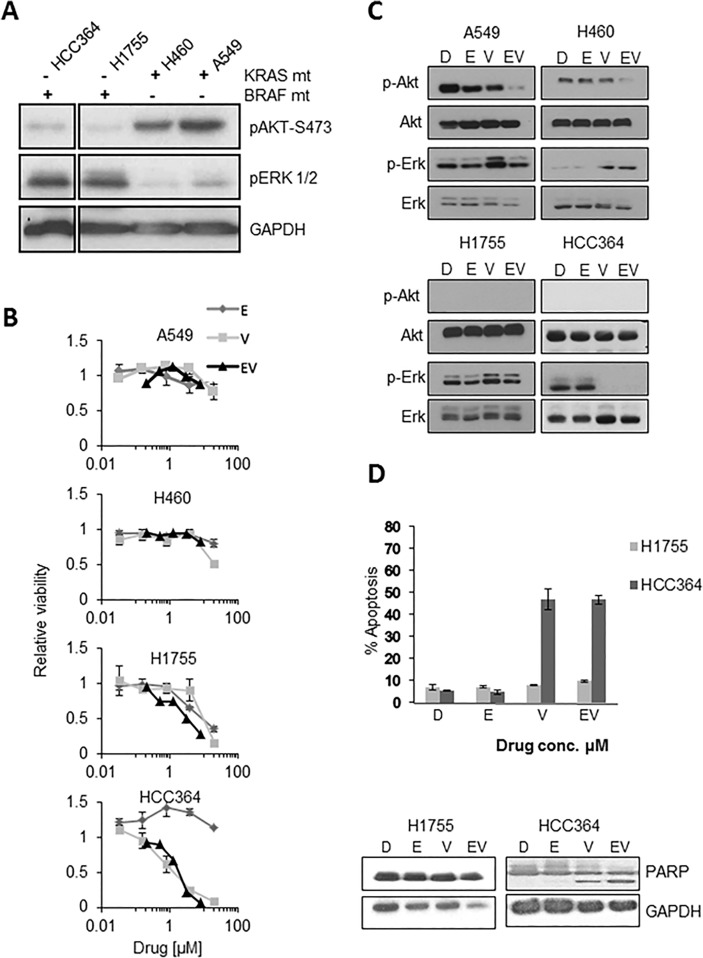
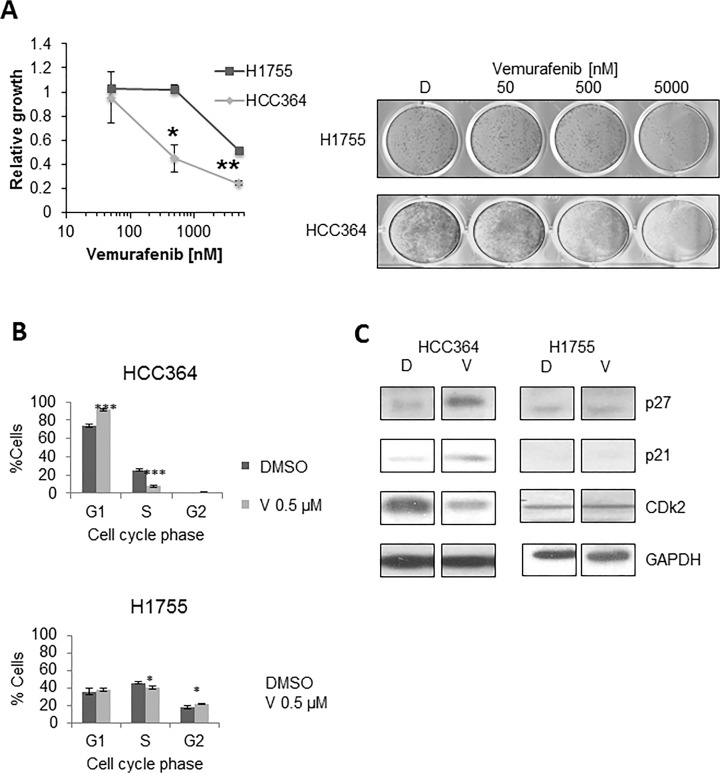
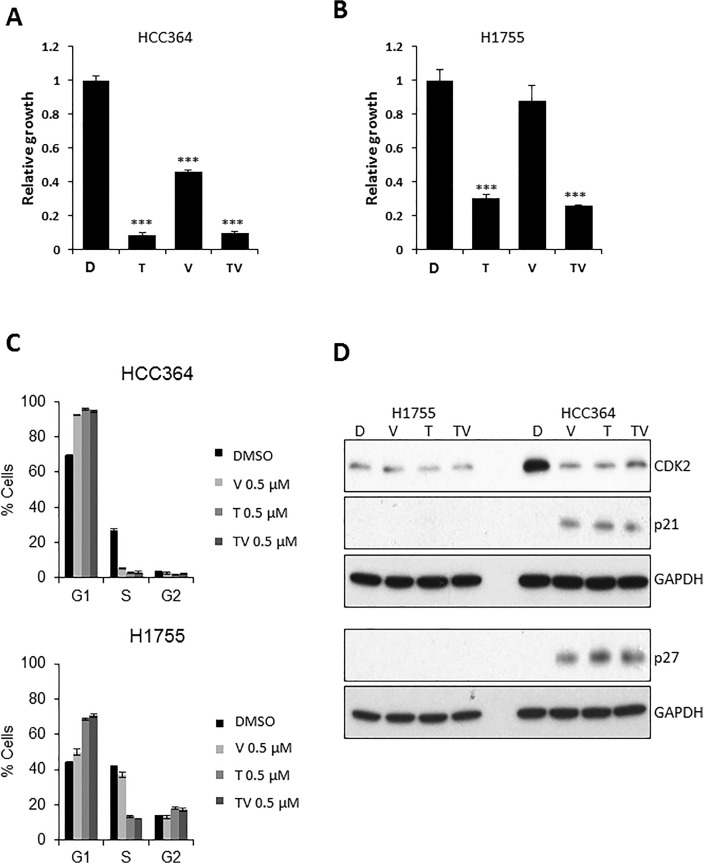
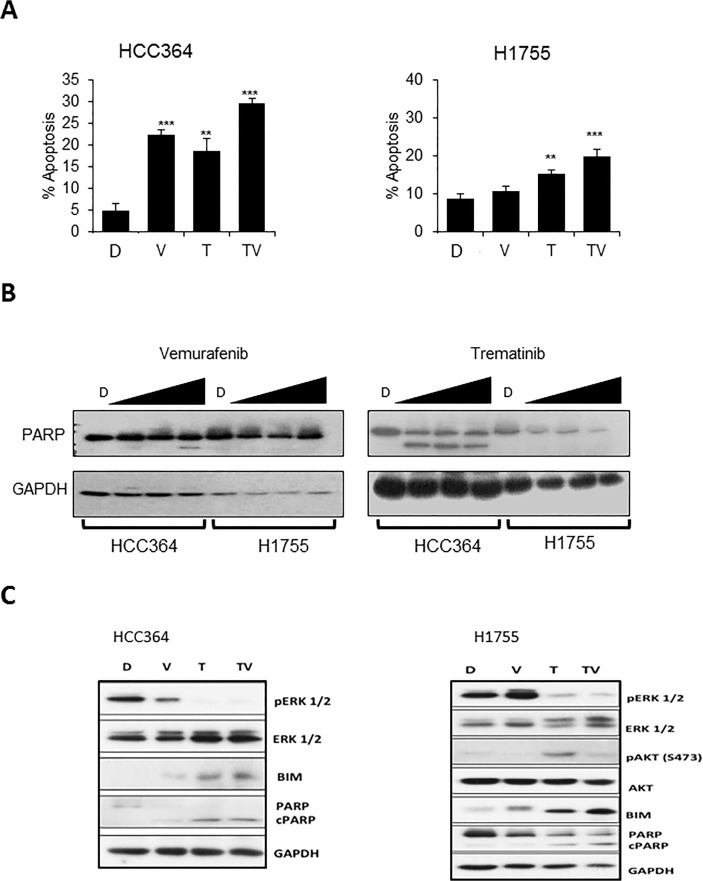
V-Raf Murine Sarcoma Viral Oncogene Homolog B (BRAF) mutated lung cancer is relatively aggressive and is resistant to currently available therapies. In a recent phase II study for patients with BRAF-V600E non-small cell lung cancer (NSCLC), BRAF V600E inhibitor demonstrated evidence of activity, but 30% of this selected group progressed while on treatment, suggesting a need for developing alternative strategies. We tested two different options to enhance the efficacy of vemurafenib (BRAF V600E inhibitor) in BRAF mutated NSCLC. The first option was the addition of erlotinib to vemurafenib to see whether the combination provided synergy. The second was to induce MEK inhibition (downstream of RAF) with trametinib (MEK inhibitor). We found that the combination of vemurafenib and erlotinib was not synergistic to the inhibition of p-ERK signaling in BRAF-V600E cells. Vemurafenib caused significant apoptosis, G1 arrest and upregulation of BIM in BRAF-V600 cells. Trametinib was effective as a single agent in BRAF mutated cells, either V600E or non-V600E. Finally, the combination of vemurafenib and trametinib caused a small but significant increase in apoptosis as well as a significant upregulation of BIM when compared to either single agent. Thus, hinting at the possibility of utilizing a combinational approach for the management of this group of patients. Importantly, trametinib alone caused upregulation of p-AKT in BRAF non-V600 mutated cells, while this effect was nullified with the combination. This finding suggests that, the combination of a MEK inhibitor with a BRAF inhibitor will be more efficacious in the clinical setting for patients with BRAF mutated NSCLC.
Conflict of interest statement
Figures
Similar articles
-
Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: clinical evidence and experience.Ther Adv Respir Dis. 2018 Jan-Dec;12:1753466618767611. doi: 10.1177/1753466618767611. Ther Adv Respir Dis. 2018. PMID: 29595366 Free PMC article. Review.
-
Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer.Cancer Res. 2012 Feb 1;72(3):779-89. doi: 10.1158/0008-5472.CAN-11-2941. Epub 2011 Dec 16. Cancer Res. 2012. PMID: 22180495
-
Comparison of dabrafenib and trametinib combination therapy with vemurafenib monotherapy on health-related quality of life in patients with unresectable or metastatic cutaneous BRAF Val600-mutation-positive melanoma (COMBI-v): results of a phase 3, open-label, randomised trial.Lancet Oncol. 2015 Oct;16(13):1389-98. doi: 10.1016/S1470-2045(15)00087-X. Lancet Oncol. 2015. PMID: 26433819 Clinical Trial.
-
Dabrafenib plus trametinib in patients with previously treated BRAF(V600E)-mutant metastatic non-small cell lung cancer: an open-label, multicentre phase 2 trial.Lancet Oncol. 2016 Jul;17(7):984-993. doi: 10.1016/S1470-2045(16)30146-2. Epub 2016 Jun 6. Lancet Oncol. 2016. PMID: 27283860 Free PMC article. Clinical Trial.
-
Therapeutic potential of trametinib to inhibit the mutagenesis by inactivating the protein kinase pathway in non-small cell lung cancer.Expert Rev Anticancer Ther. 2019 Jan;19(1):11-17. doi: 10.1080/14737140.2019.1554440. Epub 2018 Dec 4. Expert Rev Anticancer Ther. 2019. PMID: 30513023 Review.
Cited by
-
Targeted Therapies in Non-Small Cell Lung Cancer-Beyond EGFR and ALK.Cancers (Basel). 2015 May 26;7(2):930-49. doi: 10.3390/cancers7020816. Cancers (Basel). 2015. PMID: 26018876 Free PMC article. Review.
-
Targeting BRAF-Mutant Non-Small Cell Lung Cancer: From Molecular Profiling to Rationally Designed Therapy.Oncologist. 2017 Jul;22(7):786-796. doi: 10.1634/theoncologist.2016-0458. Epub 2017 May 9. Oncologist. 2017. PMID: 28487464 Free PMC article. Review.
-
[Research Progress of Targeted Therapy for BRAF Mutation in Advanced Non-small Cell Lung Cancer].Zhongguo Fei Ai Za Zhi. 2018 Aug 20;21(8):635-640. doi: 10.3779/j.issn.1009-3419.2018.08.10. Zhongguo Fei Ai Za Zhi. 2018. PMID: 30172272 Free PMC article. Review. Chinese.
-
Dabrafenib in combination with trametinib in the treatment of patients with BRAF V600-positive advanced or metastatic non-small cell lung cancer: clinical evidence and experience.Ther Adv Respir Dis. 2018 Jan-Dec;12:1753466618767611. doi: 10.1177/1753466618767611. Ther Adv Respir Dis. 2018. PMID: 29595366 Free PMC article. Review.
-
Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib.Oncotarget. 2016 Aug 26;8(36):60094-60108. doi: 10.18632/oncotarget.11635. eCollection 2017 Sep 1. Oncotarget. 2016. PMID: 28947956 Free PMC article.
References
-
- Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. - PubMed
-
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, et al. (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med 353: 123–132. - PubMed
-
- Zhou C, Wu YL, Chen G, Feng J, Liu XQ, et al. (2011) Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol 12: 735–742. 10.1016/S1470-2045(11)70184-X - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
