

An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging
- PMID: 25692570
- PMCID: PMC4334892
- DOI: 10.1371/journal.pgen.1004996
An integrative multi-scale analysis of the dynamic DNA methylation landscape in aging
Abstract
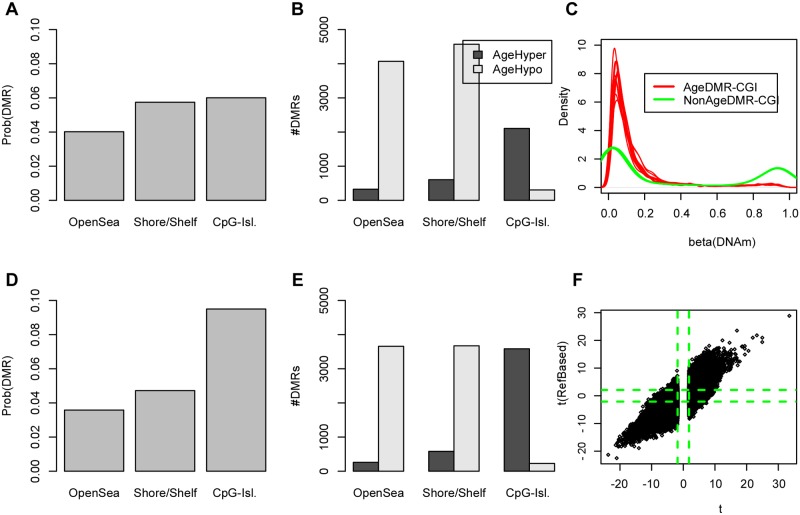
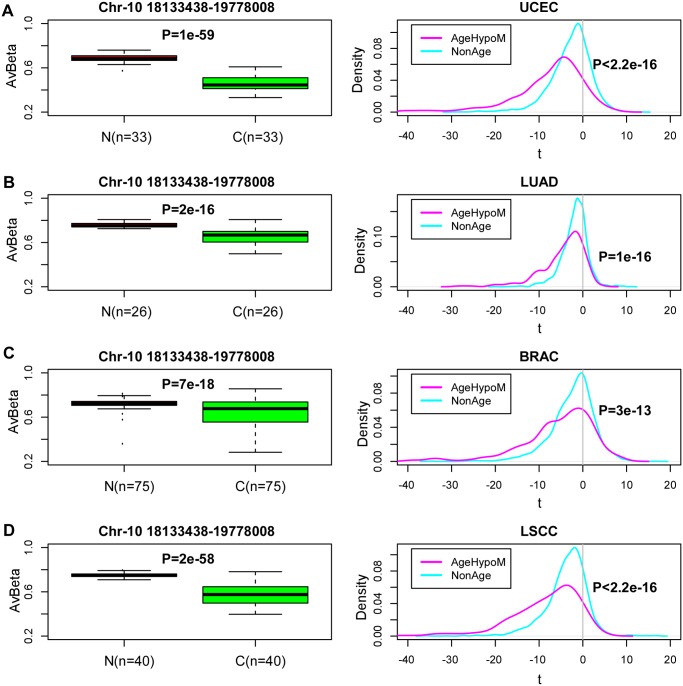
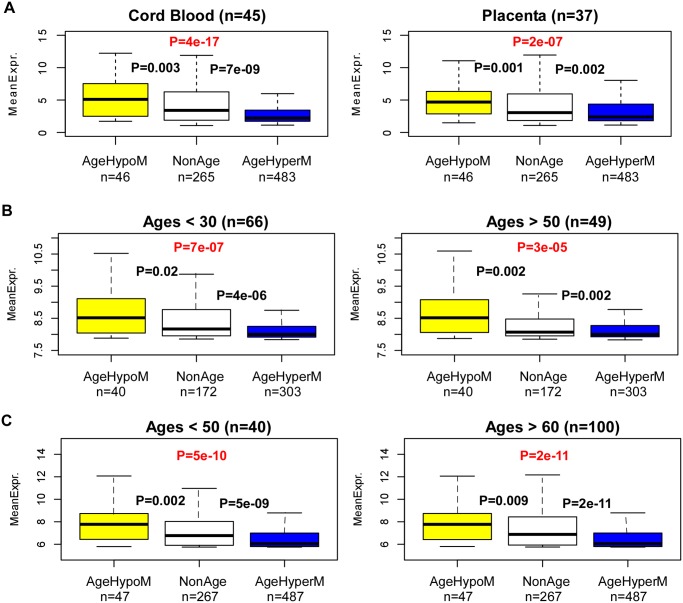
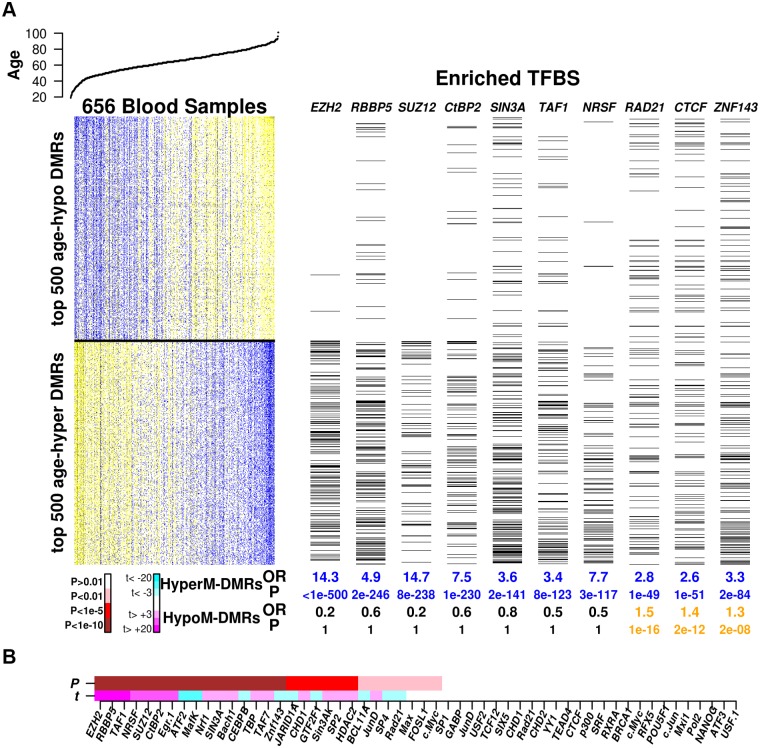
Recent studies have demonstrated that the DNA methylome changes with age. This epigenetic drift may have deep implications for cellular differentiation and disease development. However, it remains unclear how much of this drift is functional or caused by underlying changes in cell subtype composition. Moreover, no study has yet comprehensively explored epigenetic drift at different genomic length scales and in relation to regulatory elements. Here we conduct an in-depth analysis of epigenetic drift in blood tissue. We demonstrate that most of the age-associated drift is independent of the increase in the granulocyte to lymphocyte ratio that accompanies aging and that enrichment of age-hypermethylated CpG islands increases upon adjustment for cellular composition. We further find that drift has only a minimal impact on in-cis gene expression, acting primarily to stabilize pre-existing baseline expression levels. By studying epigenetic drift at different genomic length scales, we demonstrate the existence of mega-base scale age-associated hypomethylated blocks, covering approximately 14% of the human genome, and which exhibit preferential hypomethylation in age-matched cancer tissue. Importantly, we demonstrate the feasibility of integrating Illumina 450k DNA methylation with ENCODE data to identify transcription factors with key roles in cellular development and aging. Specifically, we identify REST and regulatory factors of the histone methyltransferase MLL complex, whose function may be disrupted in aging. In summary, most of the epigenetic drift seen in blood is independent of changes in blood cell type composition, and exhibits patterns at different genomic length scales reminiscent of those seen in cancer. Integration of Illumina 450k with appropriate ENCODE data may represent a fruitful approach to identify transcription factors with key roles in aging and disease.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures


Similar articles
-
Using Illumina Infinium HumanMethylation 450K BeadChip to explore genome‑wide DNA methylation profiles in a human hepatocellular carcinoma cell line.Mol Med Rep. 2018 Nov;18(5):4446-4456. doi: 10.3892/mmr.2018.9441. Epub 2018 Sep 3. Mol Med Rep. 2018. PMID: 30221710 Free PMC article.
-
Genome-wide DNA methylation analysis reveals molecular subtypes of pancreatic cancer.Oncotarget. 2017 Apr 25;8(17):28990-29012. doi: 10.18632/oncotarget.15993. Oncotarget. 2017. PMID: 28423671 Free PMC article.
-
DNA methylation and healthy human aging.Aging Cell. 2015 Dec;14(6):924-32. doi: 10.1111/acel.12349. Epub 2015 Apr 25. Aging Cell. 2015. PMID: 25913071 Free PMC article. Review.
-
Impacts of Chromatin States and Long-Range Genomic Segments on Aging and DNA Methylation.PLoS One. 2015 Jun 19;10(6):e0128517. doi: 10.1371/journal.pone.0128517. eCollection 2015. PLoS One. 2015. PMID: 26091484 Free PMC article.
-
Epigenetic Clock: Just a Convenient Marker or an Active Driver of Aging?Adv Exp Med Biol. 2019;1178:175-206. doi: 10.1007/978-3-030-25650-0_10. Adv Exp Med Biol. 2019. PMID: 31493228 Review.
Cited by
-
DNA methylation correlates of chronological age in diverse human tissue types.Epigenetics Chromatin. 2024 Aug 8;17(1):25. doi: 10.1186/s13072-024-00546-6. Epigenetics Chromatin. 2024. PMID: 39118140 Free PMC article.
-
Quantifying the stochastic component of epigenetic aging.Nat Aging. 2024 Jun;4(6):886-901. doi: 10.1038/s43587-024-00600-8. Epub 2024 May 9. Nat Aging. 2024. PMID: 38724732 Free PMC article.
-
On epigenetic stochasticity, entropy and cancer risk.Philos Trans R Soc Lond B Biol Sci. 2024 Apr 22;379(1900):20230054. doi: 10.1098/rstb.2023.0054. Epub 2024 Mar 4. Philos Trans R Soc Lond B Biol Sci. 2024. PMID: 38432318 Free PMC article. Review.
-
Transcriptional changes are tightly coupled to chromatin reorganization during cellular aging.Aging Cell. 2024 Mar;23(3):e14056. doi: 10.1111/acel.14056. Epub 2023 Dec 7. Aging Cell. 2024. PMID: 38062919 Free PMC article.
-
ATAC-clock: An aging clock based on chromatin accessibility.Geroscience. 2024 Apr;46(2):1789-1806. doi: 10.1007/s11357-023-00986-0. Epub 2023 Nov 4. Geroscience. 2024. PMID: 37924441 Free PMC article.
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
