

Lessons in de novo peptide sequencing by tandem mass spectrometry
- PMID: 25667941
- PMCID: PMC4367481
- DOI: 10.1002/mas.21406
Lessons in de novo peptide sequencing by tandem mass spectrometry
Abstract
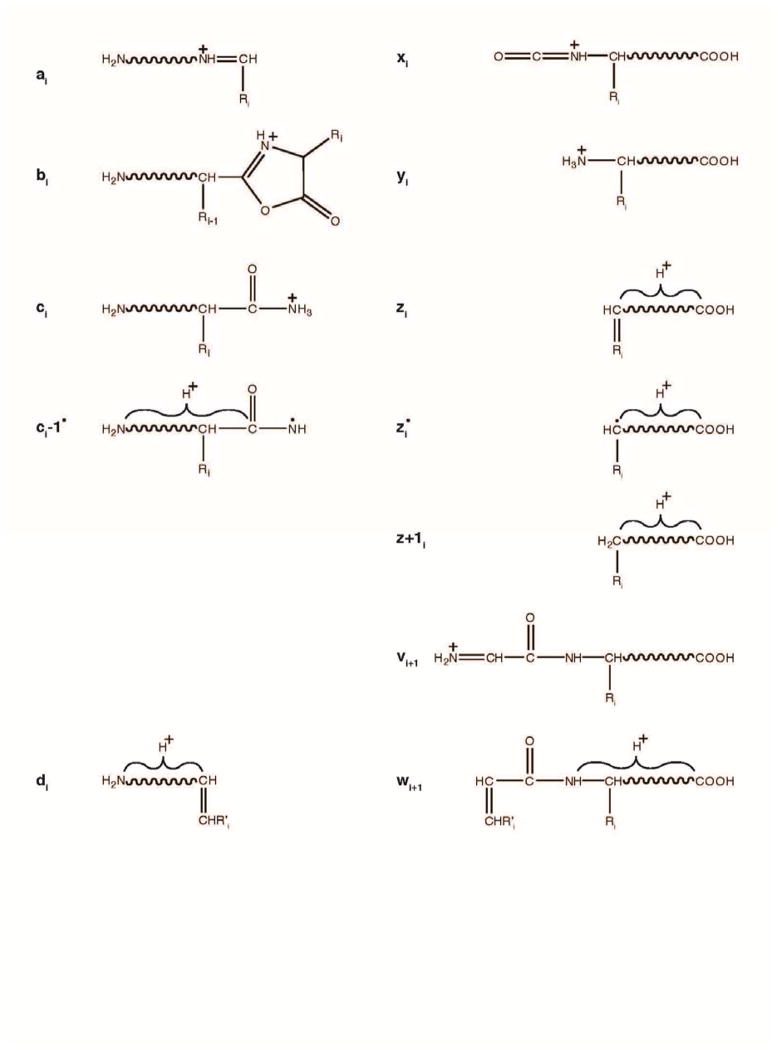
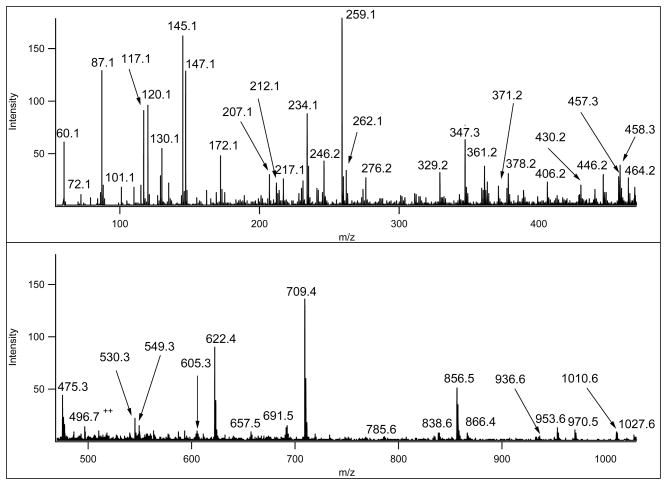
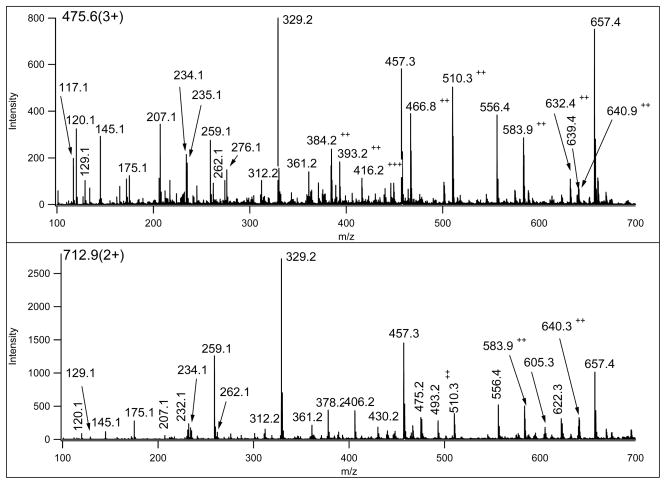
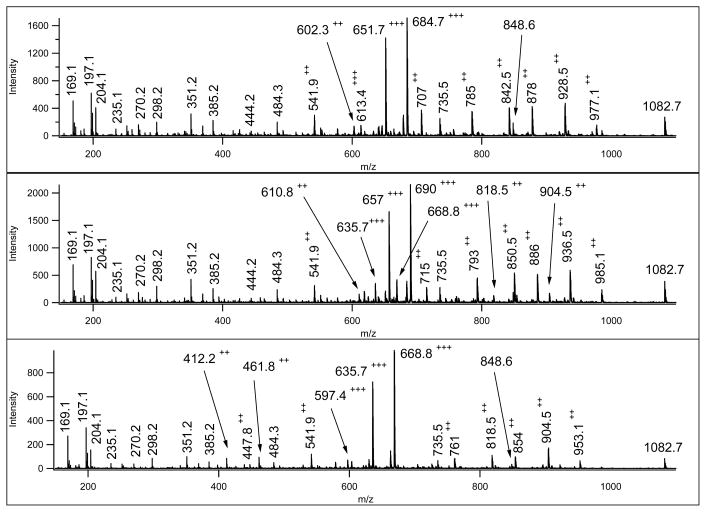
Mass spectrometry has become the method of choice for the qualitative and quantitative characterization of protein mixtures isolated from all kinds of living organisms. The raw data in these studies are MS/MS spectra, usually of peptides produced by proteolytic digestion of a protein. These spectra are "translated" into peptide sequences, normally with the help of various search engines. Data acquisition and interpretation have both been automated, and most researchers look only at the summary of the identifications without ever viewing the underlying raw data used for assignments. Automated analysis of data is essential due to the volume produced. However, being familiar with the finer intricacies of peptide fragmentation processes, and experiencing the difficulties of manual data interpretation allow a researcher to be able to more critically evaluate key results, particularly because there are many known rules of peptide fragmentation that are not incorporated into search engine scoring. Since the most commonly used MS/MS activation method is collision-induced dissociation (CID), in this article we present a brief review of the history of peptide CID analysis. Next, we provide a detailed tutorial on how to determine peptide sequences from CID data. Although the focus of the tutorial is de novo sequencing, the lessons learned and resources supplied are useful for data interpretation in general.
Figures


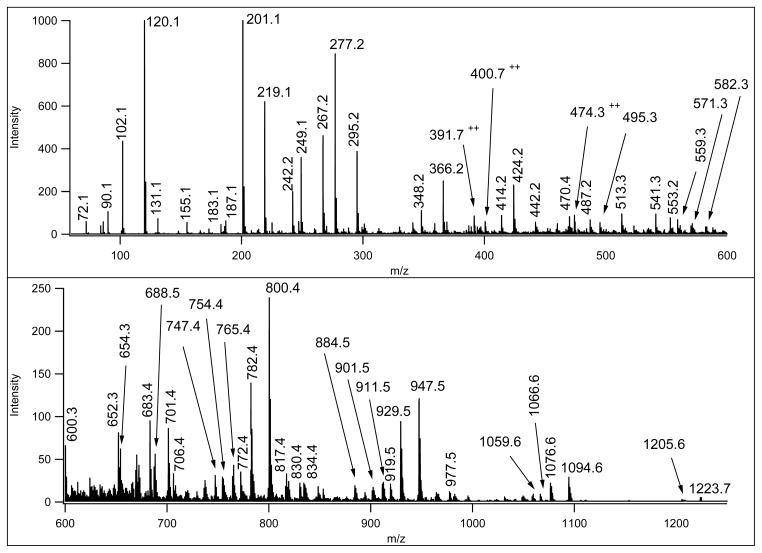

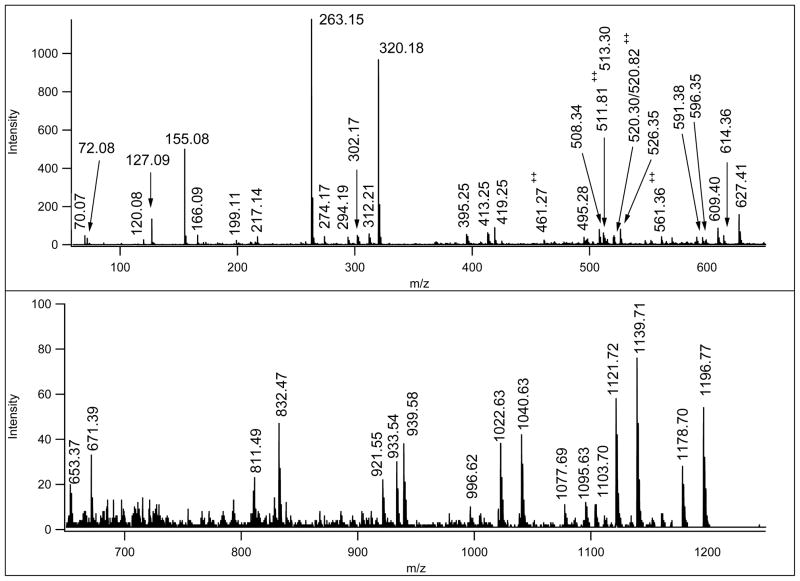
Similar articles
-
pNovo: de novo peptide sequencing and identification using HCD spectra.J Proteome Res. 2010 May 7;9(5):2713-24. doi: 10.1021/pr100182k. J Proteome Res. 2010. PMID: 20329752
-
Ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometry and tandem mass spectrometry for peptide de novo amino acid sequencing for a seven-protein mixture by paired single-residue transposed Lys-N and Lys-C digestion.Rapid Commun Mass Spectrom. 2017 Jan 30;31(2):207-217. doi: 10.1002/rcm.7783. Rapid Commun Mass Spectrom. 2017. PMID: 27813191
-
Recent Developments in Computational Methods for De Novo Peptide Sequencing from Tandem Mass Spectrometry (MS/MS).Protein Pept Lett. 2015;22(11):983-91. doi: 10.2174/0929866522666150821113127. Protein Pept Lett. 2015. PMID: 26295161
-
Protein identification by tandem mass spectrometry and sequence database searching.Methods Mol Biol. 2007;367:87-119. doi: 10.1385/1-59745-275-0:87. Methods Mol Biol. 2007. PMID: 17185772 Review.
-
Algorithms for the de novo sequencing of peptides from tandem mass spectra.Expert Rev Proteomics. 2011 Oct;8(5):645-57. doi: 10.1586/epr.11.54. Expert Rev Proteomics. 2011. PMID: 21999834 Review.
Cited by
-
RoboOligo: software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids.Nucleic Acids Res. 2015 May 26;43(10):e64. doi: 10.1093/nar/gkv145. Epub 2015 Mar 27. Nucleic Acids Res. 2015. PMID: 25820423 Free PMC article.
-
Multi-Bioactivity of Protein Digests and Peptides from Oat (Avena sativa L.) Kernels in the Prevention of the Cardiometabolic Syndrome.Molecules. 2022 Nov 15;27(22):7907. doi: 10.3390/molecules27227907. Molecules. 2022. PMID: 36432008 Free PMC article.
-
Mass Spectrometric De Novo Sequencing of Natural Peptides.Methods Mol Biol. 2024;2758:61-75. doi: 10.1007/978-1-0716-3646-6_3. Methods Mol Biol. 2024. PMID: 38549008
-
Strategies for Development of a Next-Generation Protein Sequencing Platform.Trends Biochem Sci. 2020 Jan;45(1):76-89. doi: 10.1016/j.tibs.2019.09.005. Epub 2019 Oct 30. Trends Biochem Sci. 2020. PMID: 31676211 Free PMC article. Review.
-
Discovery, characterization, and remediation of a C-terminal Fc-extension in proteins expressed in CHO cells.MAbs. 2018 Nov-Dec;10(8):1291-1300. doi: 10.1080/19420862.2018.1511197. Epub 2018 Sep 20. MAbs. 2018. PMID: 30148415 Free PMC article.
References
-
- Alexander AJ, Thibault P, Boyd RK. Collision-induced dissociation of peptide ions. 2. Remote charge-site fragmentation in a tandem, hybrid mass spectrometer. Rapid Commun Mass Spectrom. 1989;3:30–34.
-
- Bakken V, Helgaker T, Uggerud E. Models of fragmentations induced by electron attachment to protonated peptides. Eur J Mass Spectrom (Chichester, Eng) 2004;10:625–638. - PubMed
-
- Baldwin MA, Medzihradszky KF, Lock CM, Fisher B, Settineri TA, Burlingame AL. Matrix-assisted laser desorption/ionization coupled with quadrupole/orthogonal acceleration time-of-flight mass spectrometry for protein discovery, identification, and structural analysis. Anal Chem. 2001;73:1707–1720. - PubMed
-
- Ballard KD, Gaskell SJ. Sequential mass spectrometry applied to the study of the formation of “internal” fragment ions of protonated peptides. Int J Mass Spectrom Ion Processes. 1991;111:173–189.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
