

Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding
- PMID: 25597246
- PMCID: PMC4344939
- DOI: 10.1016/j.molimm.2014.12.010
Desialylation of airway epithelial cells during influenza virus infection enhances pneumococcal adhesion via galectin binding
Abstract
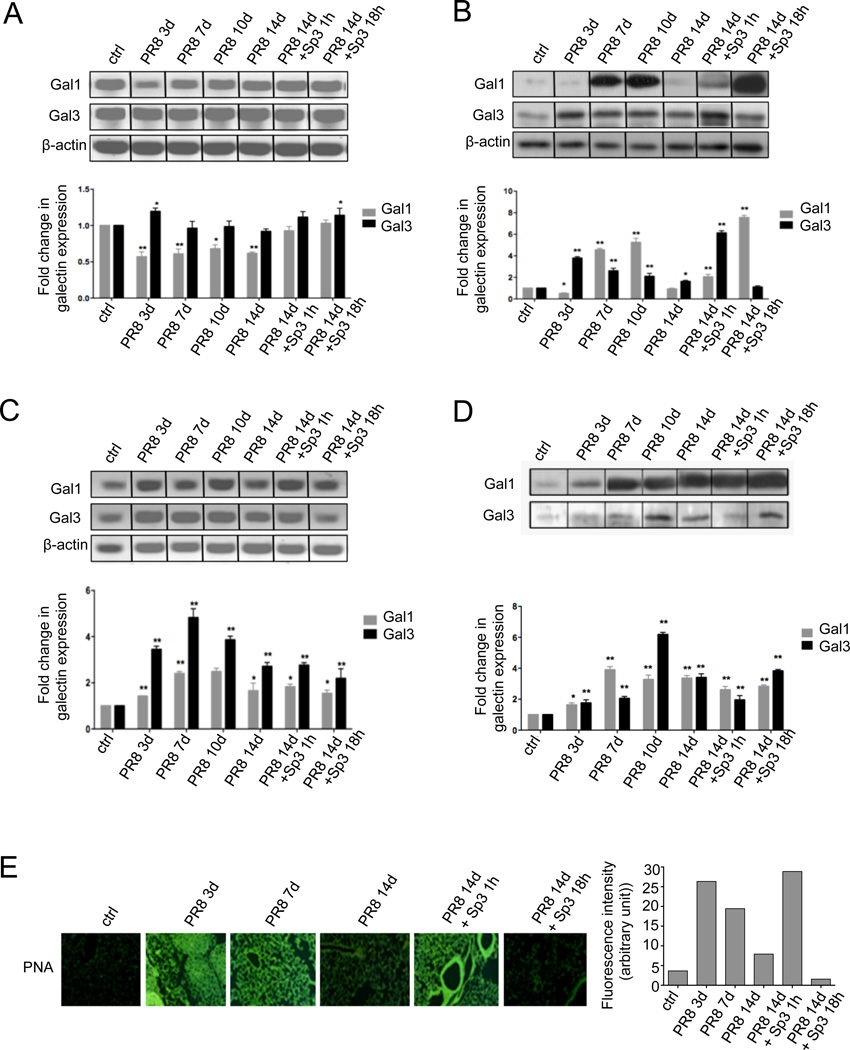
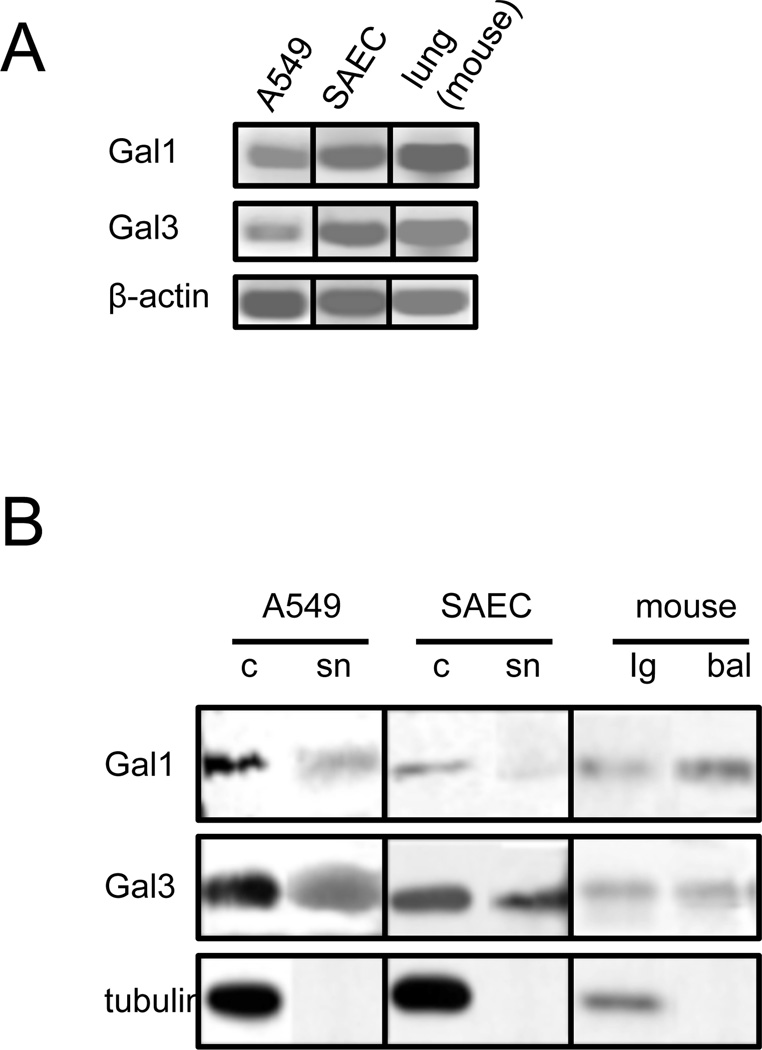
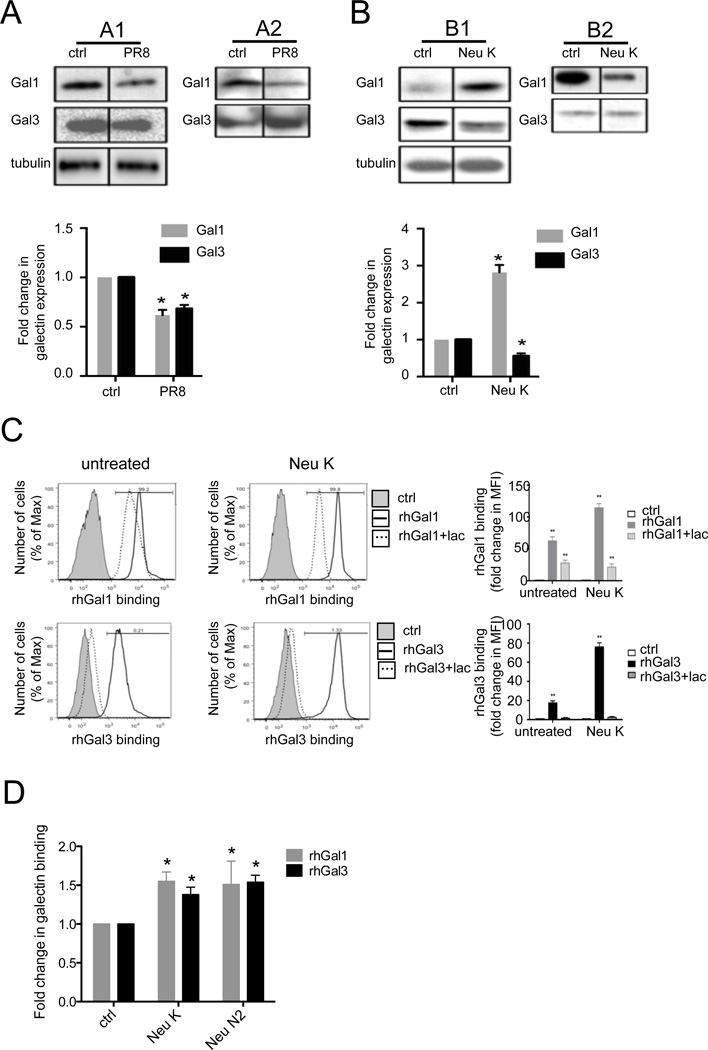
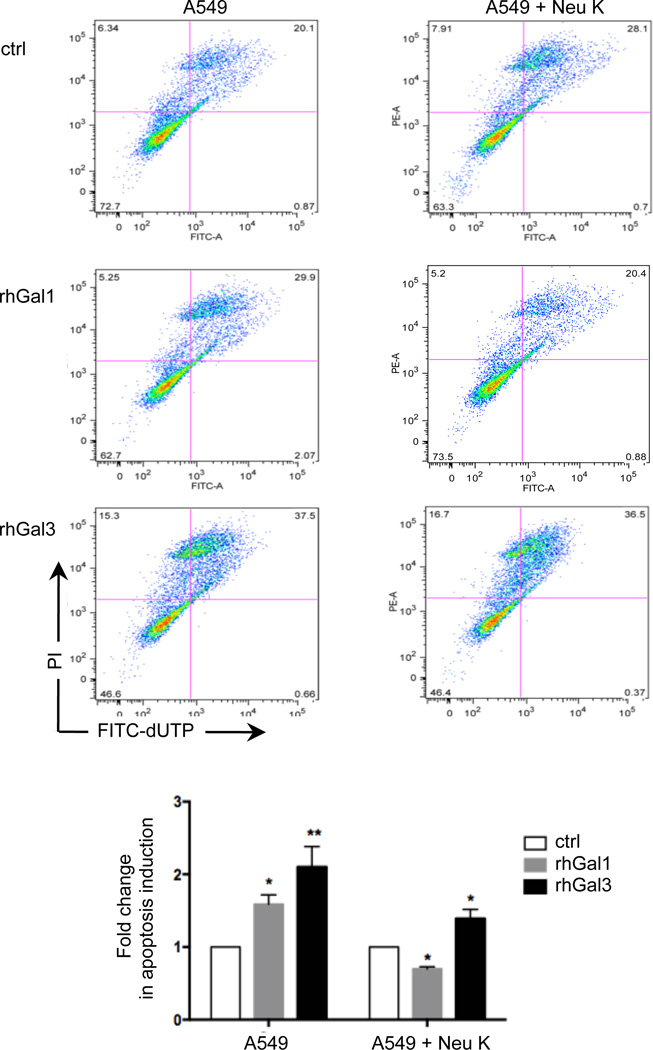
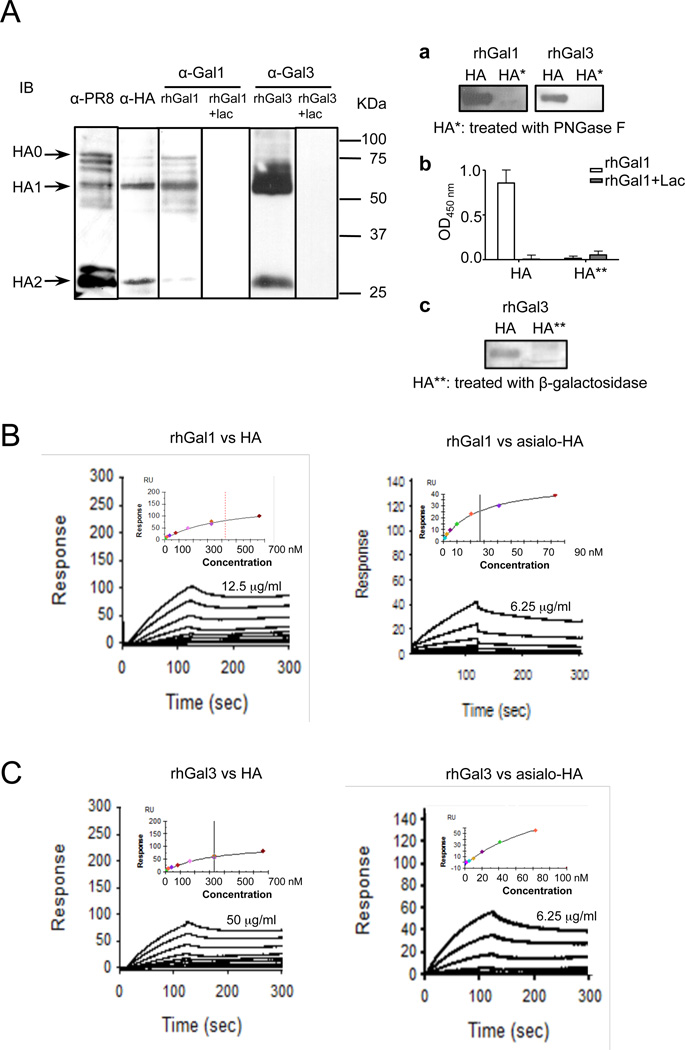
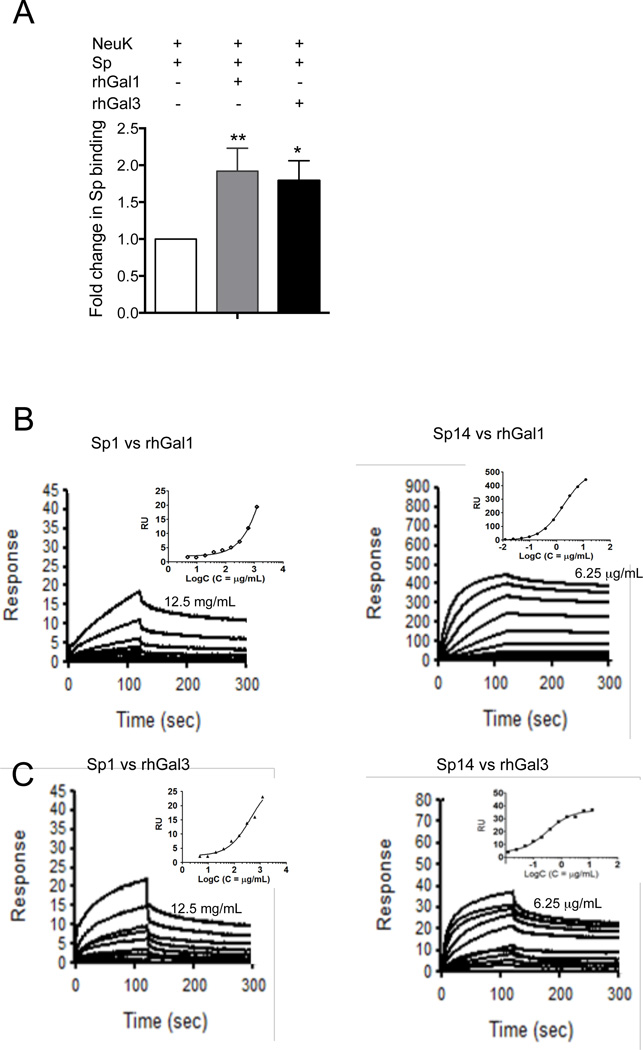
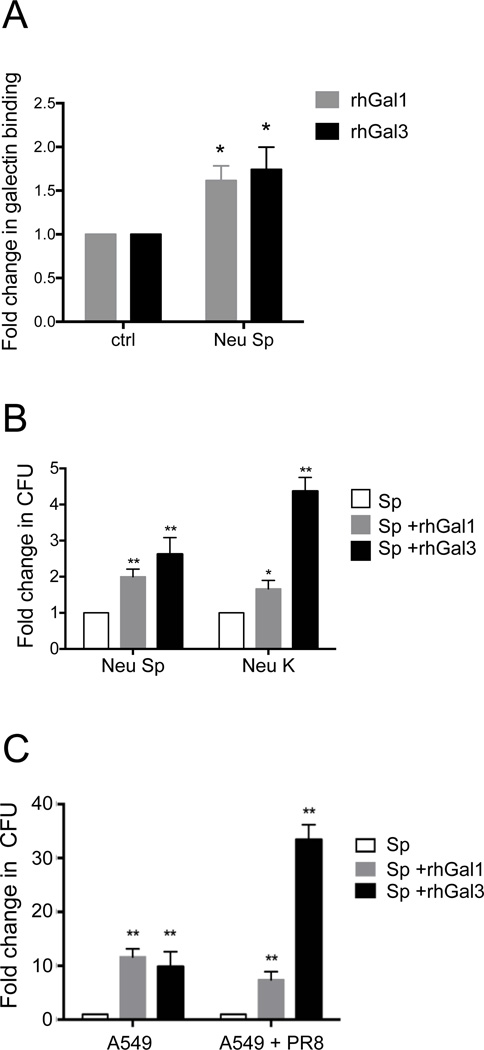
The continued threat of worldwide influenza pandemics, together with the yearly emergence of antigenically drifted influenza A virus (IAV) strains, underscore the urgent need to elucidate not only the mechanisms of influenza virulence, but also those mechanisms that predispose influenza patients to increased susceptibility to subsequent infection with Streptococcus pneumoniae. Glycans displayed on the surface of epithelia that are exposed to the external environment play important roles in microbial recognition, adhesion, and invasion. It is well established that the IAV hemagglutinin and pneumococcal adhesins enable their attachment to the host epithelia. Reciprocally, the recognition of microbial glycans by host carbohydrate-binding proteins (lectins) can initiate innate immune responses, but their relevance in influenza or pneumococcal infections is poorly understood. Galectins are evolutionarily conserved lectins characterized by affinity for β-galactosides and a unique sequence motif, with critical regulatory roles in development and immune homeostasis. In this study, we examined the possibility that galectins expressed in the airway epithelial cells might play a significant role in viral or pneumococcal adhesion to airway epithelial cells. Our results in a mouse model for influenza and pneumococcal infection revealed that the murine lung expresses a diverse galectin repertoire, from which selected galectins, including galectin 1 (Gal1) and galectin 3 (Gal3), are released to the bronchoalveolar space. Further, the results showed that influenza and subsequent S. pneumoniae infections significantly alter the glycosylation patterns of the airway epithelial surface and modulate galectin expression. In vitro studies on the human airway epithelial cell line A549 were consistent with the observations made in the mouse model, and further revealed that both Gal1 and Gal3 bind strongly to IAV and S. pneumoniae, and that exposure of the cells to viral neuraminidase or influenza infection increased galectin-mediated S. pneumoniae adhesion to the cell surface. Our results suggest that upon influenza infection, pneumococcal adhesion to the airway epithelial surface is enhanced by an interplay among the host galectins and viral and pneumococcal neuraminidases. The observed enhancement of pneumococcal adhesion may be a contributing factor to the observed hypersusceptibility to pneumonia of influenza patients.
Keywords: Airway A549 cells; Galectin; Influenza; Neuraminidase; Pneumococcus pneumoniae.
Copyright © 2014 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Galectins regulate the inflammatory response in airway epithelial cells exposed to microbial neuraminidase by modulating the expression of SOCS1 and RIG1.Mol Immunol. 2015 Dec;68(2 Pt A):194-202. doi: 10.1016/j.molimm.2015.08.005. Epub 2015 Sep 6. Mol Immunol. 2015. PMID: 26355912 Free PMC article.
-
GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection.mBio. 2021 Jun 29;12(3):e0326920. doi: 10.1128/mBio.03269-20. Epub 2021 Jun 1. mBio. 2021. PMID: 34061598 Free PMC article.
-
Influenza viral neuraminidase primes bacterial coinfection through TGF-β-mediated expression of host cell receptors.Proc Natl Acad Sci U S A. 2015 Jan 6;112(1):238-43. doi: 10.1073/pnas.1414422112. Epub 2014 Dec 22. Proc Natl Acad Sci U S A. 2015. PMID: 25535343 Free PMC article.
-
Otitis media: the chinchilla model.Microb Drug Resist. 1999 Spring;5(1):57-72. doi: 10.1089/mdr.1999.5.57. Microb Drug Resist. 1999. PMID: 10332723 Review.
-
Impact of pneumococcal microbial surface components recognizing adhesive matrix molecules on colonization.Mol Oral Microbiol. 2012 Aug;27(4):246-56. doi: 10.1111/j.2041-1014.2012.00654.x. Epub 2012 Jun 15. Mol Oral Microbiol. 2012. PMID: 22759310 Review.
Cited by
-
Hyperinflammation and Fibrosis in Severe COVID-19 Patients: Galectin-3, a Target Molecule to Consider.Front Immunol. 2020 Aug 18;11:2069. doi: 10.3389/fimmu.2020.02069. eCollection 2020. Front Immunol. 2020. PMID: 32973815 Free PMC article.
-
Exploration of Galectin Ligands Displayed on Gram-Negative Respiratory Bacterial Pathogens with Different Cell Surface Architectures.Biomolecules. 2021 Apr 18;11(4):595. doi: 10.3390/biom11040595. Biomolecules. 2021. PMID: 33919637 Free PMC article.
-
Galectin-1 mediates interactions between polymorphonuclear leukocytes and vascular endothelial cells, and promotes their extravasation during lipopolysaccharide-induced acute lung injury.Mol Immunol. 2023 Apr;156:127-135. doi: 10.1016/j.molimm.2023.02.011. Epub 2023 Mar 13. Mol Immunol. 2023. PMID: 36921487 Free PMC article.
-
Interrupting the Conversation: Implications for Crosstalk Between Viral and Bacterial Infections in the Asthmatic Airway.Front Allergy. 2021 Oct 26;2:738987. doi: 10.3389/falgy.2021.738987. eCollection 2021. Front Allergy. 2021. PMID: 35386999 Free PMC article. Review.
-
Galectin-3: A Harbinger of Reactive Oxygen Species, Fibrosis, and Inflammation in Pulmonary Arterial Hypertension.Antioxid Redox Signal. 2019 Nov 10;31(14):1053-1069. doi: 10.1089/ars.2019.7753. Epub 2019 Mar 29. Antioxid Redox Signal. 2019. PMID: 30767565 Free PMC article. Review.
References
-
- Ahmad N, Gabius HJ, et al. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J Biol Chem. 2004;279(12):10841–10847. - PubMed
-
- Barker WH, Mullooly JP. Impact of epidemic type A influenza in a defined adult population. Am J Epidemiol. 1980;112(6):798–811. - PubMed
-
- Barker WH, Mullooly JP. Pneumonia and influenza deaths during epidemics: implications for prevention. Arch Intern Med. 1982;142(1):85–89. - PubMed
-
- Barrionuevo P, Beigier-Bompadre M, et al. A novel function for galectin-1 at the crossroad of innate and adaptive immunity: galectin-1 regulates monocyte/macrophage physiology through a nonapoptotic ERK-dependent pathway. J Immunol. 2007;178(1):436–445. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
