

Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3
- PMID: 25590633
- PMCID: PMC4295939
- DOI: 10.1371/journal.pgen.1004834
Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3
Abstract
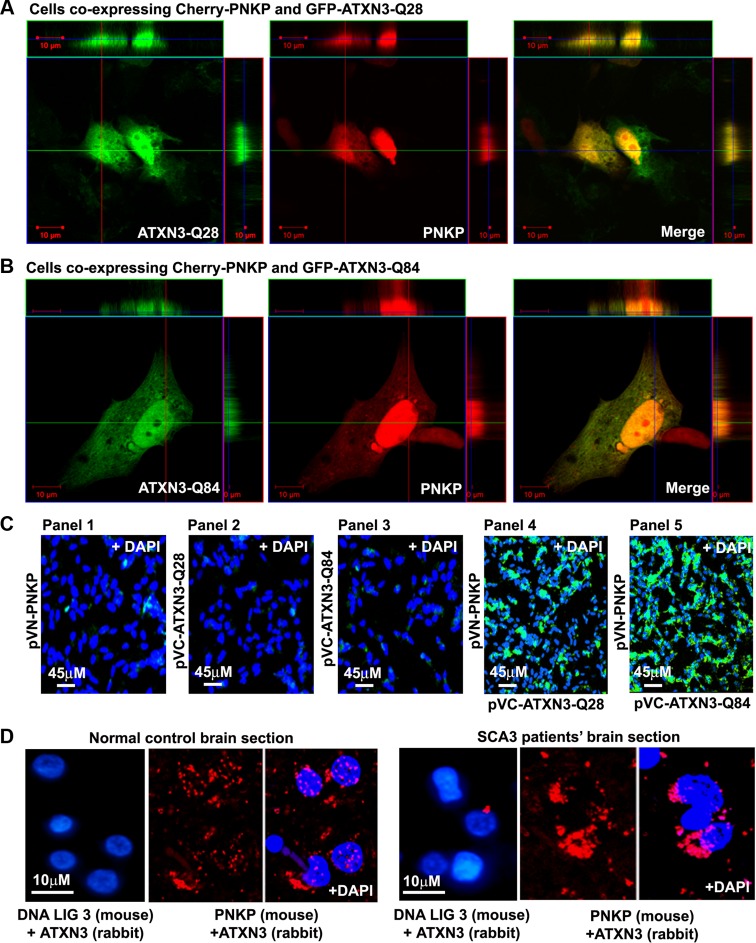
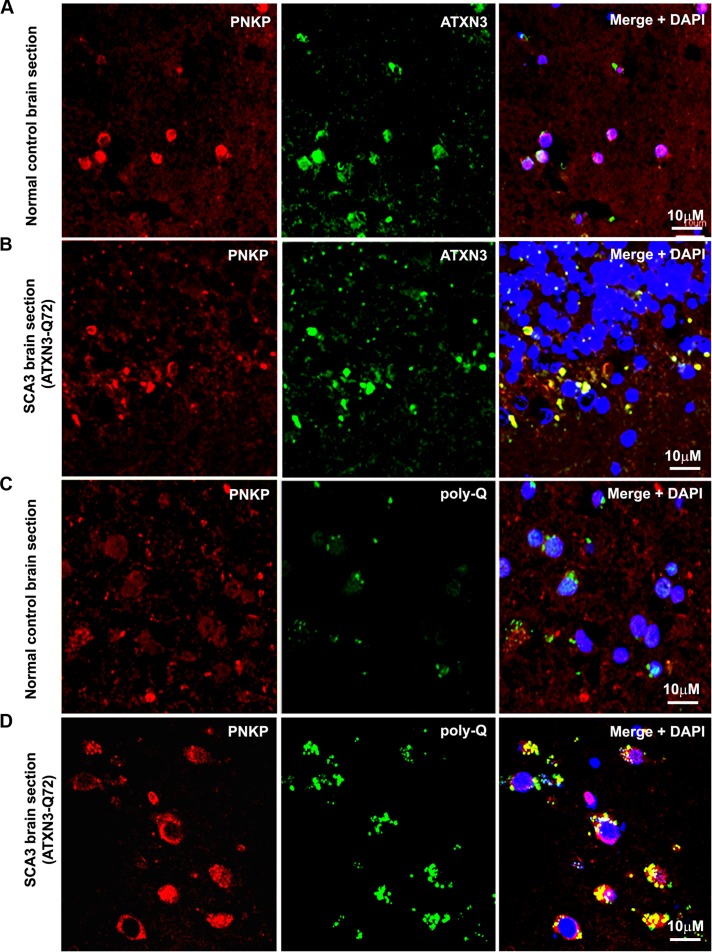
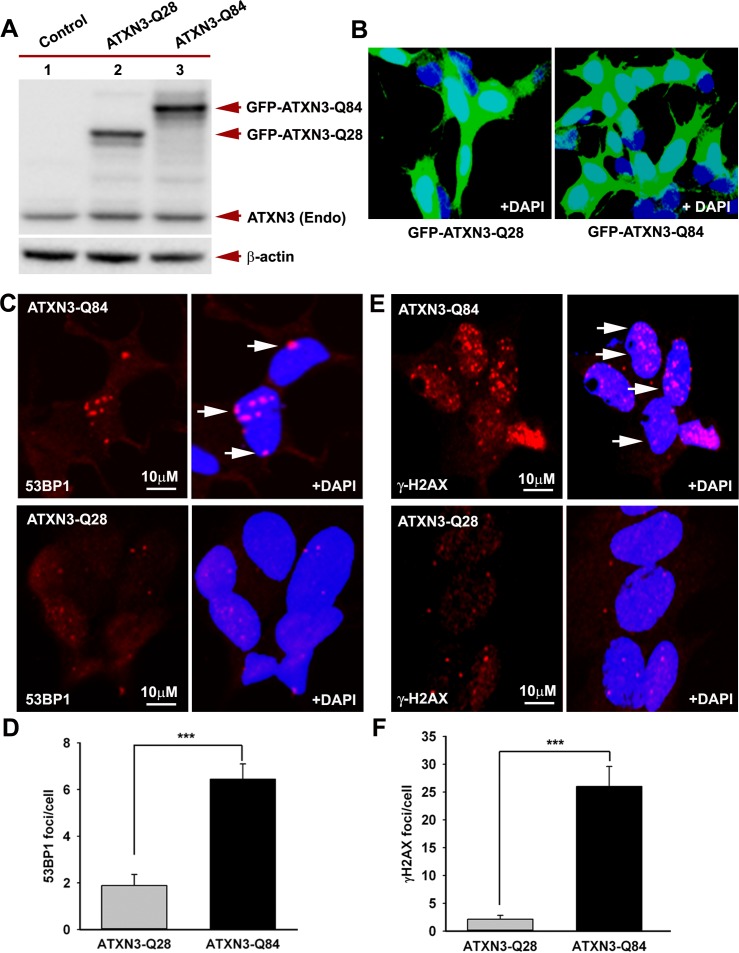
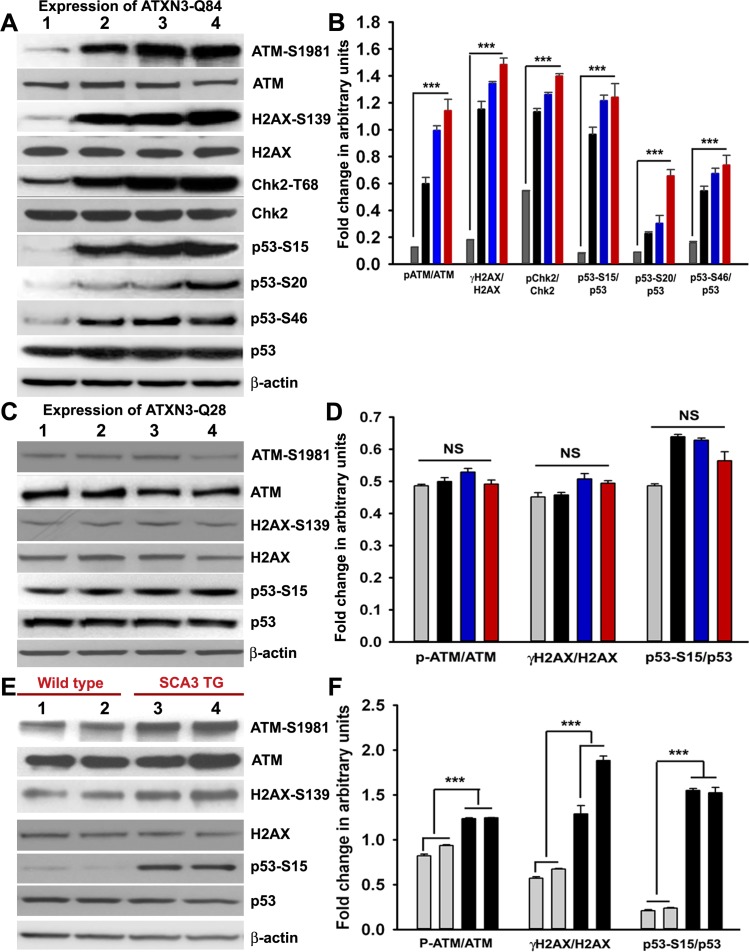
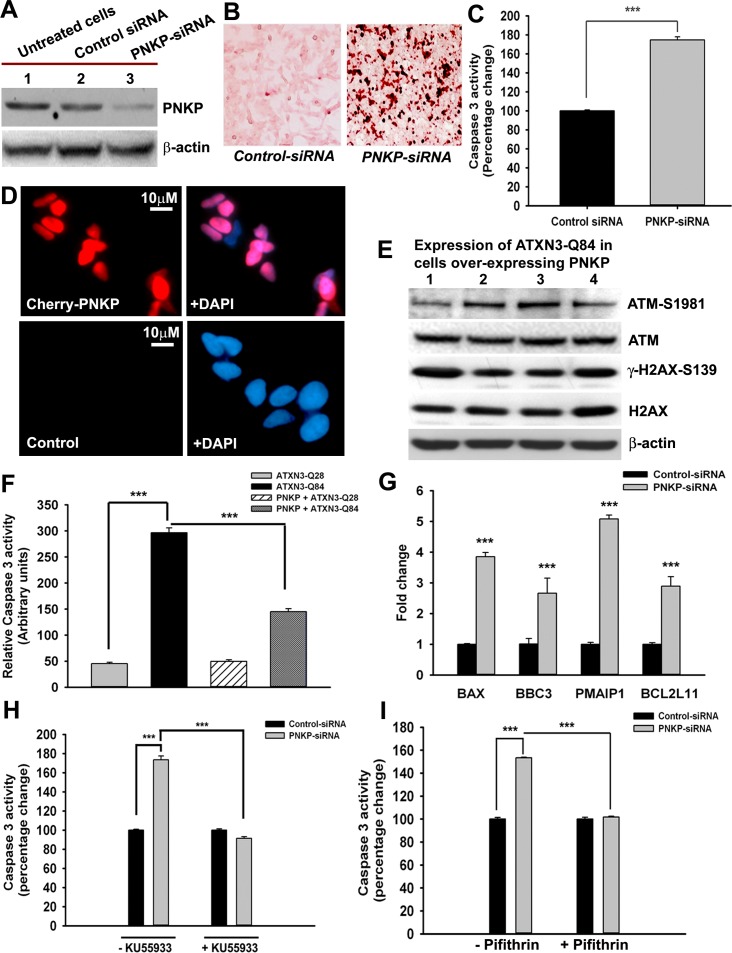
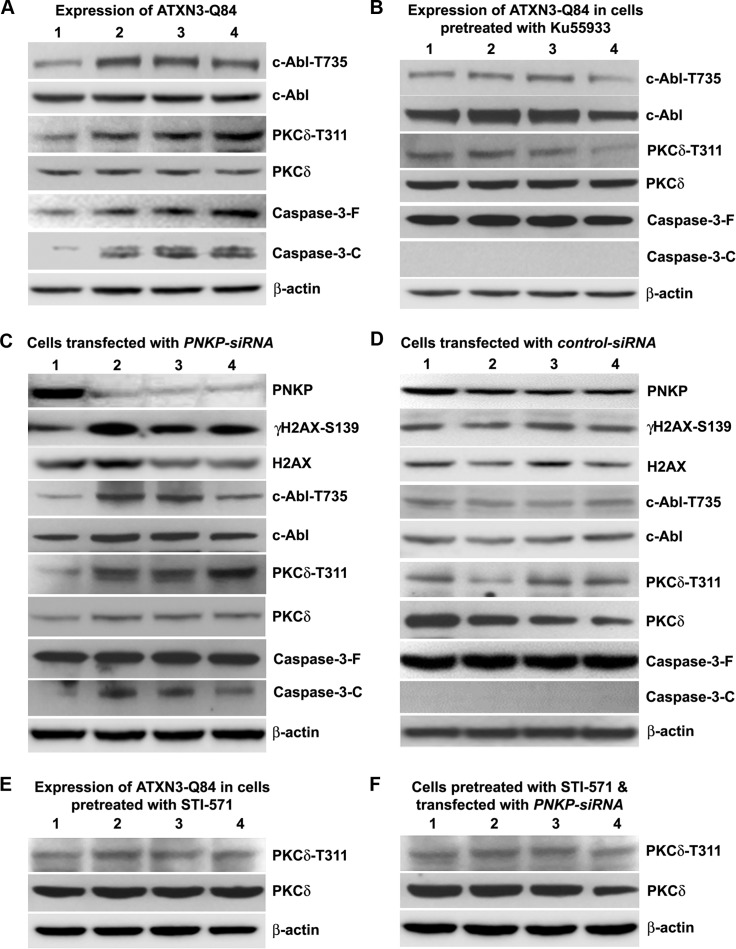
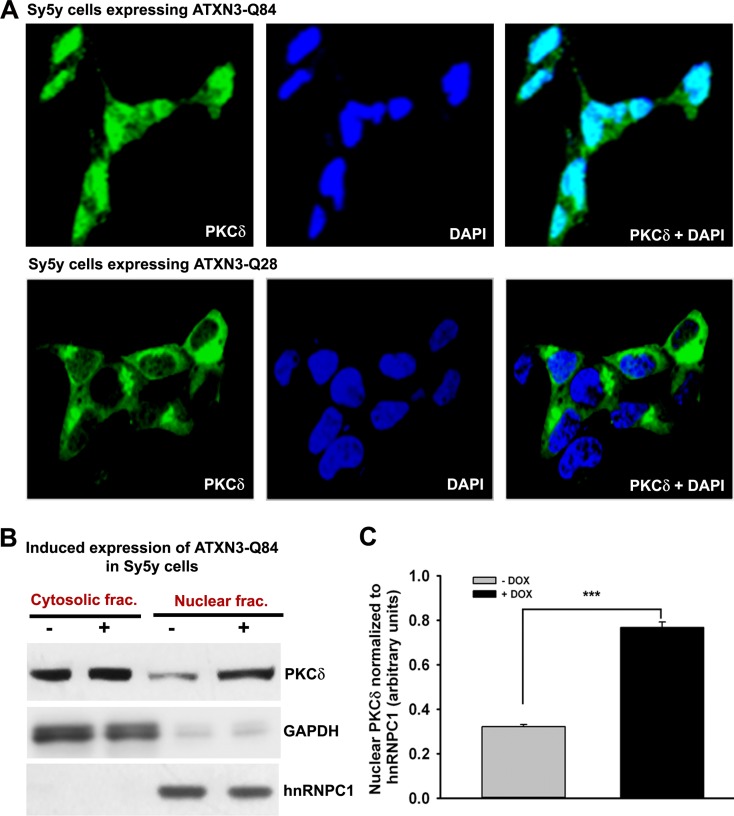
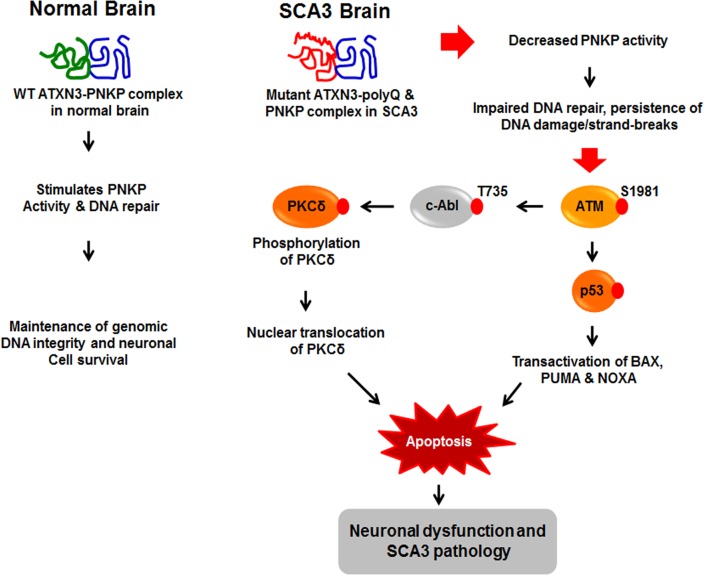
Spinocerebellar ataxia type 3 (SCA3), also known as Machado-Joseph disease (MJD), is an untreatable autosomal dominant neurodegenerative disease, and the most common such inherited ataxia worldwide. The mutation in SCA3 is the expansion of a polymorphic CAG tri-nucleotide repeat sequence in the C-terminal coding region of the ATXN3 gene at chromosomal locus 14q32.1. The mutant ATXN3 protein encoding expanded glutamine (polyQ) sequences interacts with multiple proteins in vivo, and is deposited as aggregates in the SCA3 brain. A large body of literature suggests that the loss of function of the native ATNX3-interacting proteins that are deposited in the polyQ aggregates contributes to cellular toxicity, systemic neurodegeneration and the pathogenic mechanism in SCA3. Nonetheless, a significant understanding of the disease etiology of SCA3, the molecular mechanism by which the polyQ expansions in the mutant ATXN3 induce neurodegeneration in SCA3 has remained elusive. In the present study, we show that the essential DNA strand break repair enzyme PNKP (polynucleotide kinase 3'-phosphatase) interacts with, and is inactivated by, the mutant ATXN3, resulting in inefficient DNA repair, persistent accumulation of DNA damage/strand breaks, and subsequent chronic activation of the DNA damage-response ataxia telangiectasia-mutated (ATM) signaling pathway in SCA3. We report that persistent accumulation of DNA damage/strand breaks and chronic activation of the serine/threonine kinase ATM and the downstream p53 and protein kinase C-δ pro-apoptotic pathways trigger neuronal dysfunction and eventually neuronal death in SCA3. Either PNKP overexpression or pharmacological inhibition of ATM dramatically blocked mutant ATXN3-mediated cell death. Discovery of the mechanism by which mutant ATXN3 induces DNA damage and amplifies the pro-death signaling pathways provides a molecular basis for neurodegeneration due to PNKP inactivation in SCA3, and for the first time offers a possible approach to treatment.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Comment in
-
Ataxin-3, DNA damage repair, and SCA3 cerebellar degeneration: on the path to parsimony?PLoS Genet. 2015 Jan 29;11(1):e1004937. doi: 10.1371/journal.pgen.1004937. eCollection 2015 Jan. PLoS Genet. 2015. PMID: 25633989 Free PMC article. No abstract available.
Similar articles
-
The role of the mammalian DNA end-processing enzyme polynucleotide kinase 3'-phosphatase in spinocerebellar ataxia type 3 pathogenesis.PLoS Genet. 2015 Jan 29;11(1):e1004749. doi: 10.1371/journal.pgen.1004749. eCollection 2015 Jan. PLoS Genet. 2015. PMID: 25633985 Free PMC article.
-
Deficiency in classical nonhomologous end-joining-mediated repair of transcribed genes is linked to SCA3 pathogenesis.Proc Natl Acad Sci U S A. 2020 Apr 7;117(14):8154-8165. doi: 10.1073/pnas.1917280117. Epub 2020 Mar 23. Proc Natl Acad Sci U S A. 2020. PMID: 32205441 Free PMC article.
-
A fine balance between Prpf19 and Exoc7 in achieving degradation of aggregated protein and suppression of cell death in spinocerebellar ataxia type 3.Cell Death Dis. 2021 Feb 2;12(2):136. doi: 10.1038/s41419-021-03444-x. Cell Death Dis. 2021. PMID: 33542212 Free PMC article.
-
Toward understanding Machado-Joseph disease.Prog Neurobiol. 2012 May;97(2):239-57. doi: 10.1016/j.pneurobio.2011.11.006. Epub 2011 Nov 23. Prog Neurobiol. 2012. PMID: 22133674 Free PMC article. Review.
-
Machado-Joseph disease/spinocerebellar ataxia type 3.Handb Clin Neurol. 2012;103:437-49. doi: 10.1016/B978-0-444-51892-7.00027-9. Handb Clin Neurol. 2012. PMID: 21827905 Free PMC article. Review.
Cited by
-
Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3.Sci Rep. 2016 Oct 12;6:35200. doi: 10.1038/srep35200. Sci Rep. 2016. PMID: 27731380 Free PMC article.
-
Ataxin-3 promotes genome integrity by stabilizing Chk1.Nucleic Acids Res. 2017 May 5;45(8):4532-4549. doi: 10.1093/nar/gkx095. Nucleic Acids Res. 2017. PMID: 28180282 Free PMC article.
-
Crosstalk between Different DNA Repair Pathways Contributes to Neurodegenerative Diseases.Biology (Basel). 2021 Feb 19;10(2):163. doi: 10.3390/biology10020163. Biology (Basel). 2021. PMID: 33669593 Free PMC article. Review.
-
Pathogenetic Mechanisms Underlying Spinocerebellar Ataxia Type 3 Are Altered in Primary Oligodendrocyte Culture.Cells. 2022 Aug 22;11(16):2615. doi: 10.3390/cells11162615. Cells. 2022. PMID: 36010688 Free PMC article.
-
The central role of DNA damage and repair in CAG repeat diseases.Dis Model Mech. 2018 Jan 30;11(1):dmm031930. doi: 10.1242/dmm.031930. Dis Model Mech. 2018. PMID: 29419417 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
