

mTORC2 modulates feedback regulation of p38 MAPK activity via DUSP10/MKP5 to confer differential responses to PP242 in glioblastoma
- PMID: 25568665
- PMCID: PMC4279437
- DOI: 10.18632/genesandcancer.41
mTORC2 modulates feedback regulation of p38 MAPK activity via DUSP10/MKP5 to confer differential responses to PP242 in glioblastoma
Abstract
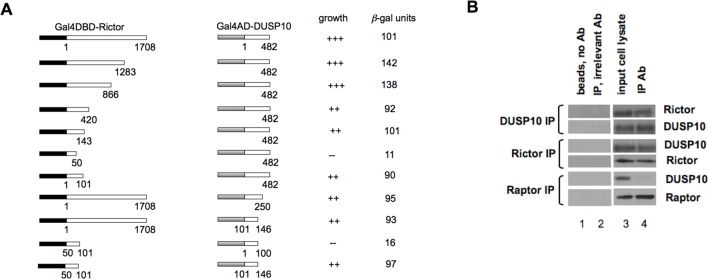
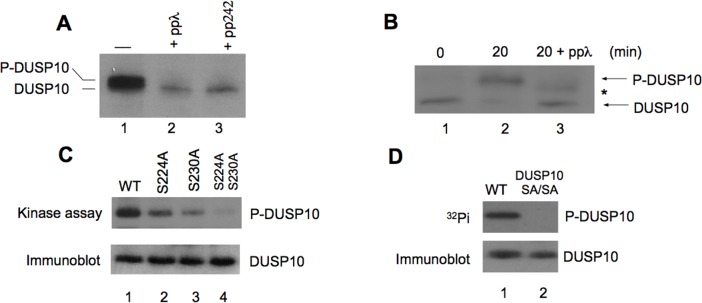
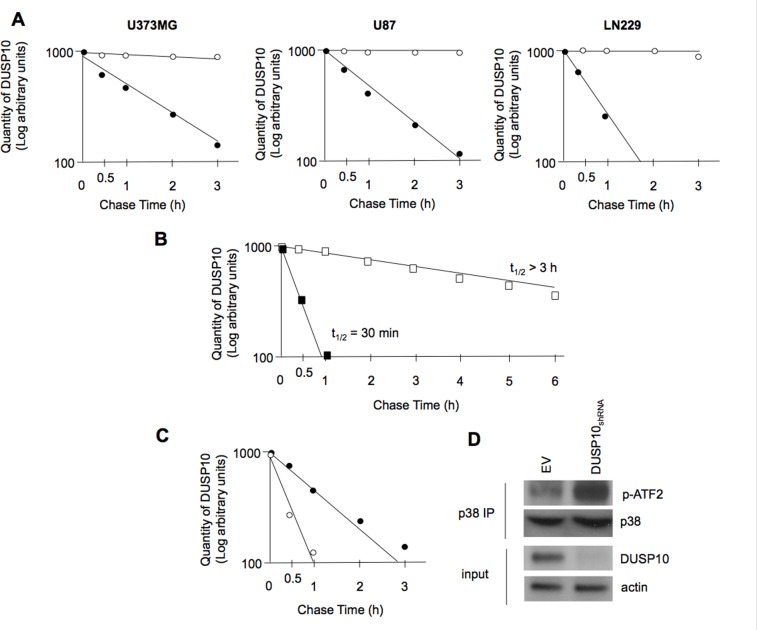
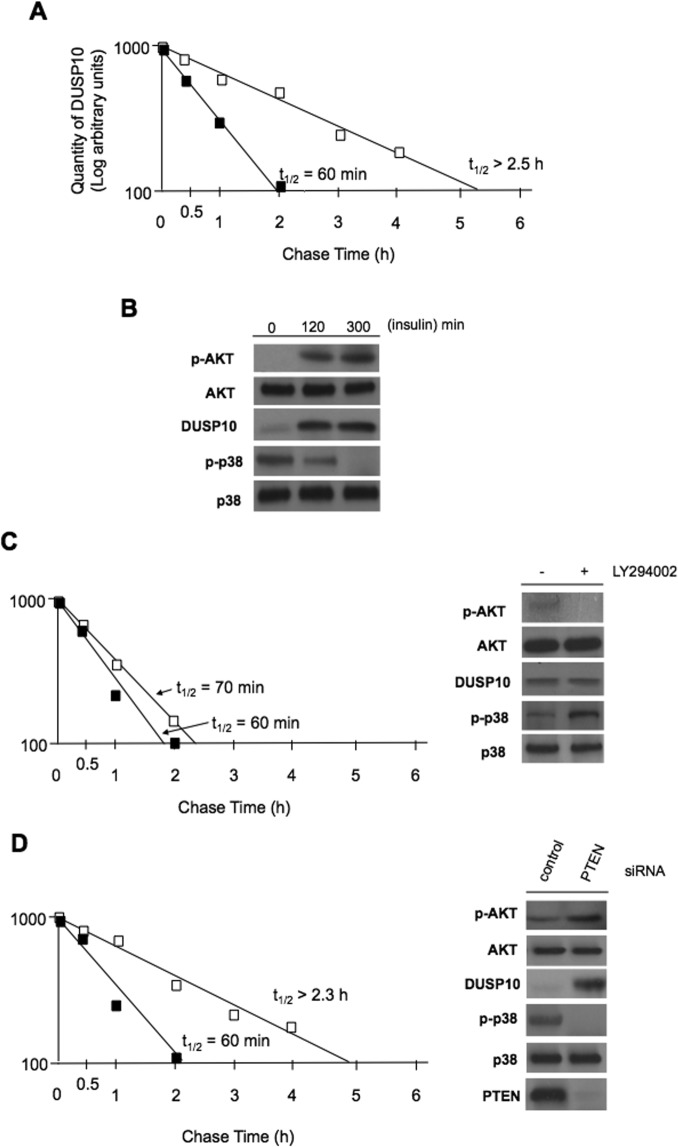
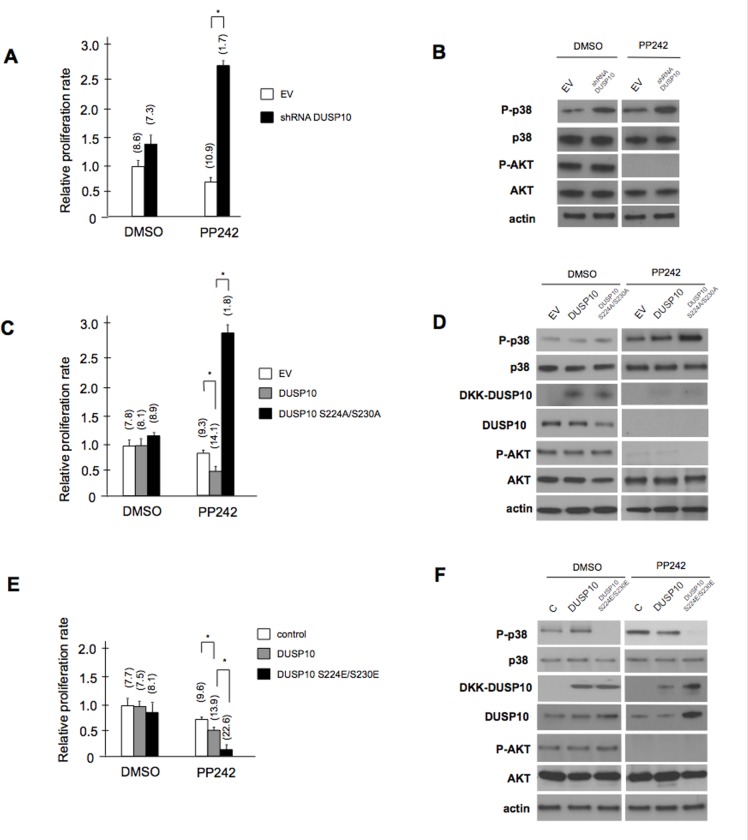
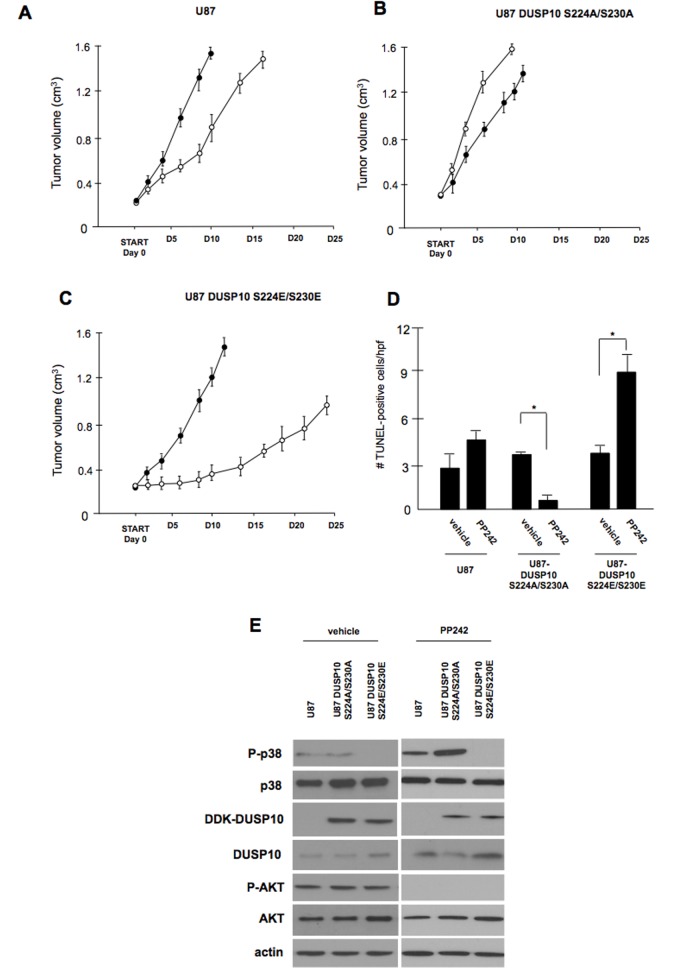
Dual-specificity phosphatases (DUSPs) dephosphorylate MAP kinases (MAPKs) resulting in their inactivation. Activation of MAPK signaling leads to enhanced DUSP expression, thus establishing feedback regulation of the MAPK pathway. The DUSPs are subject to regulation at the post-translational level via phosphorylation resulting in alterations of protein stability. Here we report that mTORC2 function leads to stabilization of the p38 MAPK phosphatase, DUSP10, thereby inhibiting p38 activity. We demonstrate that mTORC2 binds DUSP10 and phosphorylates DUSP10 on serine residues 224 and 230. These phosphorylation events block DUSP10 turnover resulting in inactivation of p38 signaling. We further show that insulin-stimulated PI3K/mTORC2 signaling regulates DUSP10 stability and p38 activity. Importantly, knockdown of DUSP10 or ectopic overexpression of nonphosphorylatable or phosphomimetic DUSP10 mutants was sufficient to confer differential mTOR kinase inhibitor responses to GBM cells in vitro and in murine xenografts. Finally, DUSP10 was shown to be overexpressed in a significant number of GBM patients. These data demonstrate the ability of the mTORC2 pathway to exert regulatory effects on the DUSP10/p38 feedback loop to control the cellular effects of mTOR kinase inhibitors in GBM and support the use of DUSP10 expression as a surrogate biomarker to predict responsiveness.
Keywords: DUSP; mTOR; p38 MAPK.
Figures
Similar articles
-
Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability.Int J Mol Sci. 2019 May 30;20(11):2668. doi: 10.3390/ijms20112668. Int J Mol Sci. 2019. PMID: 31151270 Free PMC article. Review.
-
High molecular weight hyaluronic acid regulates MMP13 expression in chondrocytes via DUSP10/MKP5.J Orthop Res. 2017 Feb;35(2):331-339. doi: 10.1002/jor.23266. Epub 2016 Apr 29. J Orthop Res. 2017. PMID: 27101204
-
Dual-specificity phosphatase 10 controls brown adipocyte differentiation by modulating the phosphorylation of p38 mitogen-activated protein kinase.PLoS One. 2013 Aug 20;8(8):e72340. doi: 10.1371/journal.pone.0072340. eCollection 2013. PLoS One. 2013. PMID: 23977283 Free PMC article.
-
The Dual-Specificity Phosphatase 10 (DUSP10): Its Role in Cancer, Inflammation, and Immunity.Int J Mol Sci. 2019 Apr 1;20(7):1626. doi: 10.3390/ijms20071626. Int J Mol Sci. 2019. PMID: 30939861 Free PMC article. Review.
-
p38 MAPK activation through B7-H3-mediated DUSP10 repression promotes chemoresistance.Sci Rep. 2019 Apr 9;9(1):5839. doi: 10.1038/s41598-019-42303-w. Sci Rep. 2019. PMID: 30967582 Free PMC article.
Cited by
-
Specific blockade of Rictor-mTOR association inhibits mTORC2 activity and is cytotoxic in glioblastoma.PLoS One. 2017 Apr 28;12(4):e0176599. doi: 10.1371/journal.pone.0176599. eCollection 2017. PLoS One. 2017. Retraction in: PLoS One. 2023 Sep 8;18(9):e0291490. doi: 10.1371/journal.pone.0291490. PMID: 28453552 Free PMC article. Retracted.
-
Rapamycin promotes differentiation increasing βIII-tubulin, NeuN, and NeuroD while suppressing nestin expression in glioblastoma cells.Oncotarget. 2017 May 2;8(18):29574-29599. doi: 10.18632/oncotarget.15906. Oncotarget. 2017. PMID: 28418837 Free PMC article.
-
Mechanistic Target of Rapamycin (mTOR) Inhibition Synergizes with Reduced Internal Ribosome Entry Site (IRES)-mediated Translation of Cyclin D1 and c-MYC mRNAs to Treat Glioblastoma.J Biol Chem. 2016 Jul 1;291(27):14146-14159. doi: 10.1074/jbc.M116.726927. Epub 2016 May 11. J Biol Chem. 2016. PMID: 27226604 Free PMC article.
-
Phosphorylation of the Hippo Pathway Component AMOTL2 by the mTORC2 Kinase Promotes YAP Signaling, Resulting in Enhanced Glioblastoma Growth and Invasiveness.J Biol Chem. 2015 Aug 7;290(32):19387-401. doi: 10.1074/jbc.M115.656587. Epub 2015 May 21. J Biol Chem. 2015. PMID: 25998128 Free PMC article.
-
Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability.Int J Mol Sci. 2019 May 30;20(11):2668. doi: 10.3390/ijms20112668. Int J Mol Sci. 2019. PMID: 31151270 Free PMC article. Review.
References
-
- Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270(13):7420–7426. - PubMed
-
- Todd JL, Tanner KG, Denu JM. Extracellular regulated kinases (ERK) 1 and ERK2 are authentic substrates for the dual-specificity protein-tyrosine phosphatase VHR. A novel role in down-regulating the ERK pathway. J Biol Chem. 1999;274(19):13271–13280. - PubMed
-
- Brondello JM, Pouyssegur J, McKenzie FR. Reduced MAP kinase phosphatase-1 degradation after p42/p44MAPK-dependent phosphorylation. Science. 1999;286(5449):2514–2517. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
