

Ataxia telangiectasia mutated kinase mediates NF-κB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection
- PMID: 25520509
- PMCID: PMC4325710
- DOI: 10.1128/JVI.02458-14
Ataxia telangiectasia mutated kinase mediates NF-κB serine 276 phosphorylation and interferon expression via the IRF7-RIG-I amplification loop in paramyxovirus infection
Abstract
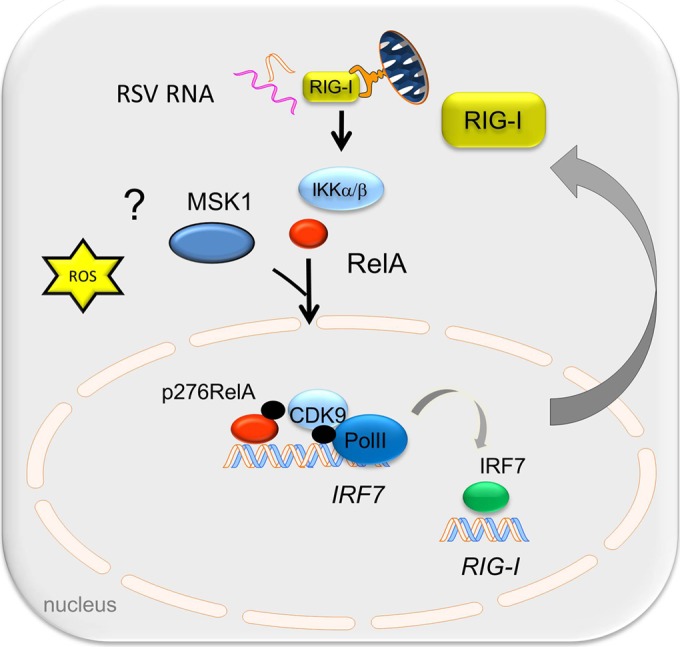
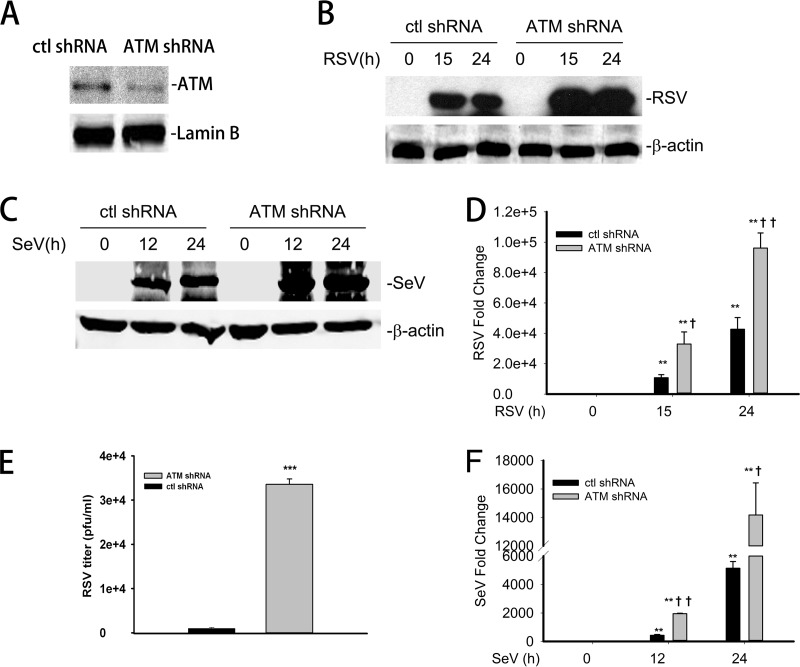
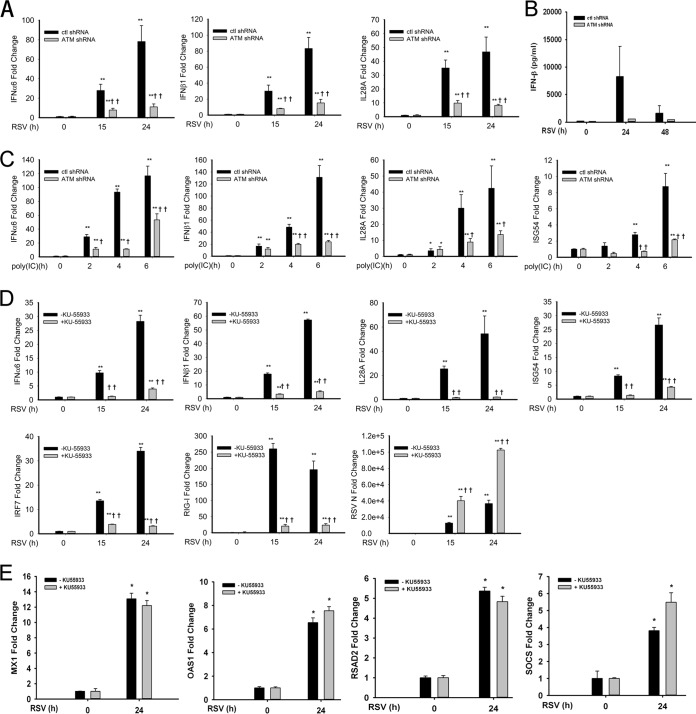
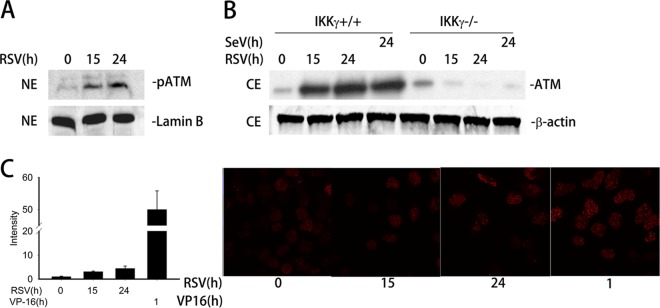
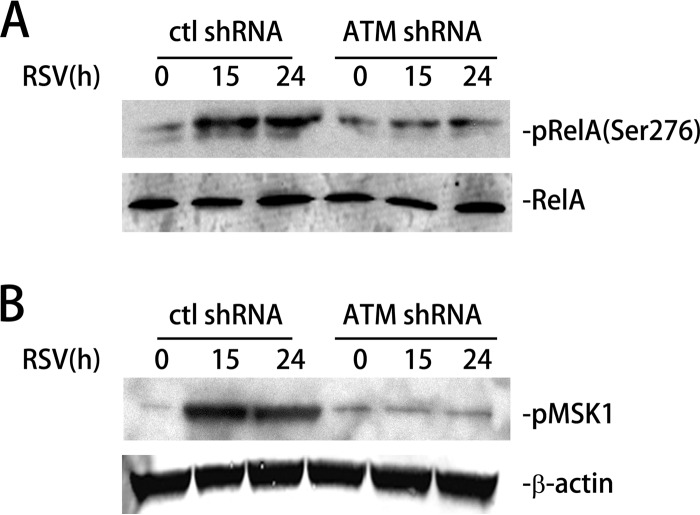
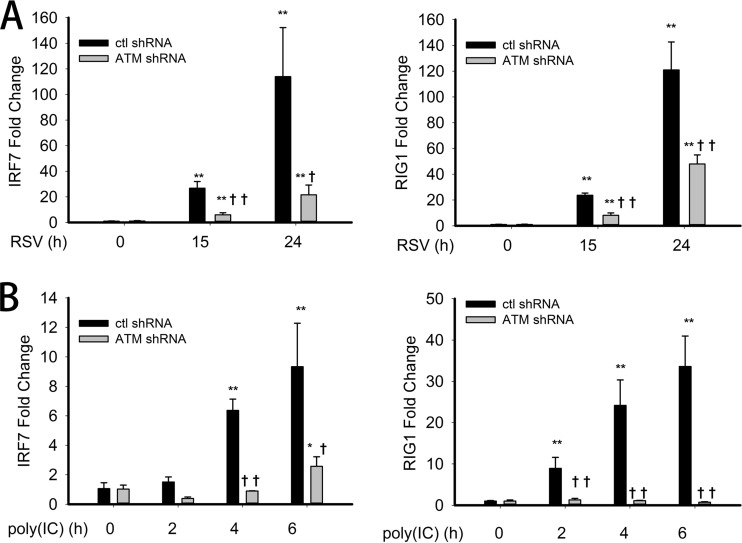
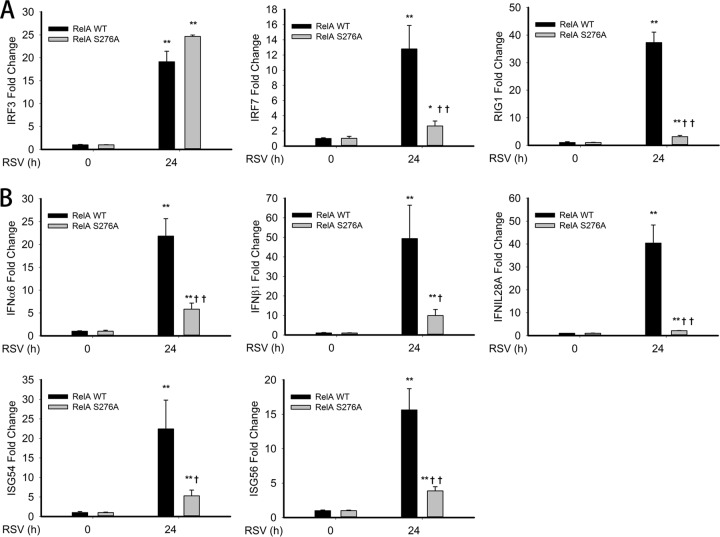
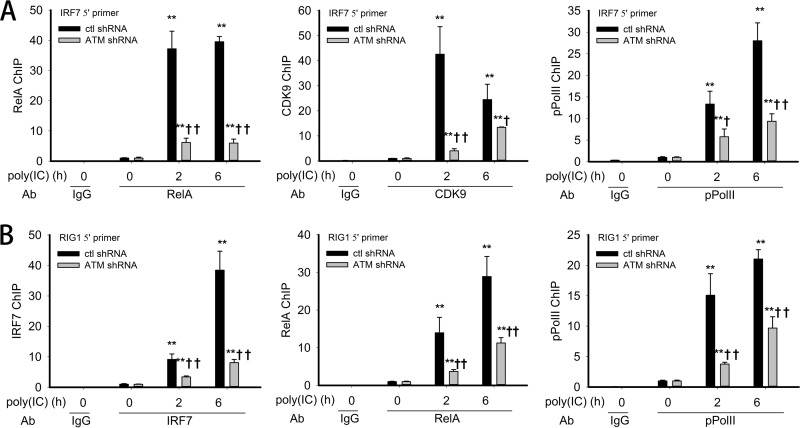
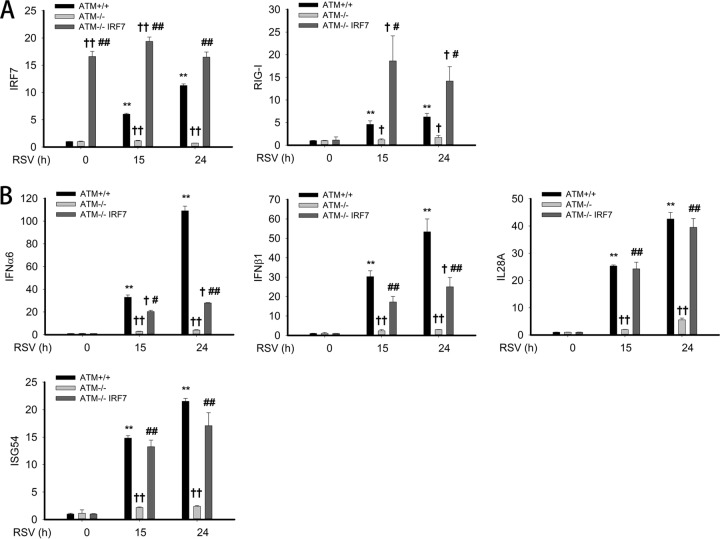
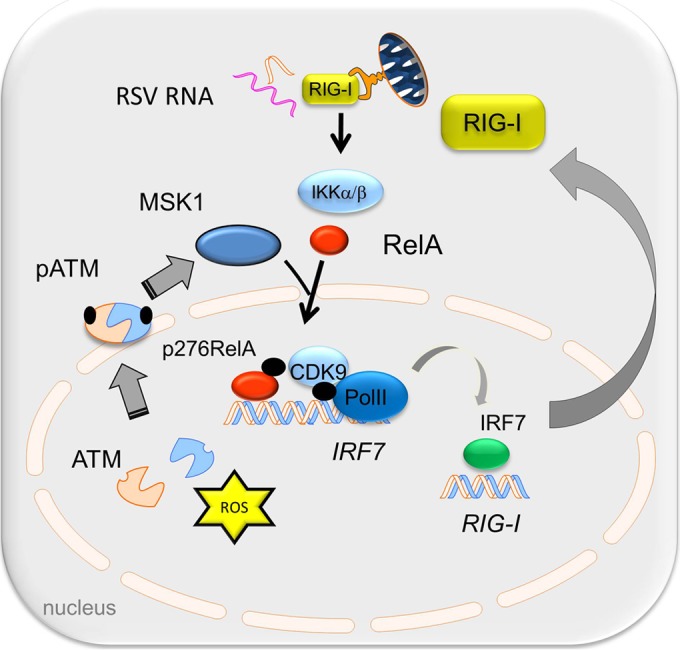
Respiratory syncytial virus (RSV) is a primary etiological agent of childhood lower respiratory tract disease. Molecular patterns induced by active infection trigger a coordinated retinoic acid-inducible gene I (RIG-I)-Toll-like receptor (TLR) signaling response to induce inflammatory cytokines and antiviral mucosal interferons. Recently, we discovered a nuclear oxidative stress-sensitive pathway mediated by the DNA damage response protein, ataxia telangiectasia mutated (ATM), in cytokine-induced NF-κB/RelA Ser 276 phosphorylation. Here we observe that ATM silencing results in enhanced single-strand RNA (ssRNA) replication of RSVand Sendai virus, due to decreased expression and secretion of type I and III interferons (IFNs), despite maintenance of IFN regulatory factor 3 (IRF3)-dependent IFN-stimulated genes (ISGs). In addition to enhanced oxidative stress, RSV replication enhances foci of phosphorylated histone 2AX variant (γH2AX), Ser 1981 phosphorylation of ATM, and IKKγ/NEMO-dependent ATM nuclear export, indicating activation of the DNA damage response. ATM-deficient cells show defective RSV-induced mitogen and stress-activated kinase 1 (MSK-1) Ser 376 phosphorylation and reduced RelA Ser 276 phosphorylation, whose formation is required for IRF7 expression. We observe that RelA inducibly binds the native IFN regulatory factor 7 (IRF7) promoter in an ATM-dependent manner, and IRF7 inducibly binds to the endogenous retinoic acid-inducible gene I (RIG-I) promoter. Ectopic IRF7 expression restores RIG-I expression and type I/III IFN expression in ATM-silenced cells. We conclude that paramyxoviruses trigger the DNA damage response, a pathway required for MSK1 activation of phospho Ser 276 RelA formation to trigger the IRF7-RIG-I amplification loop necessary for mucosal IFN production. These data provide the molecular pathogenesis for defects in the cellular innate immunity of patients with homozygous ATM mutations.
Importance: RNA virus infections trigger cellular response pathways to limit spread to adjacent tissues. This "innate immune response" is mediated by germ line-encoded pattern recognition receptors that trigger activation of two, largely independent, intracellular NF-κB and IRF3 transcription factors. Downstream, expression of protective antiviral interferons is amplified by positive-feedback loops mediated by inducible interferon regulatory factors (IRFs) and retinoic acid inducible gene (RIG-I). Our results indicate that a nuclear oxidative stress- and DNA damage-sensing factor, ATM, is required to mediate a cross talk pathway between NF-κB and IRF7 through mediating phosphorylation of NF-κB. Our studies provide further information about the defects in cellular and innate immunity in patients with inherited ATM mutations.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
Figures
Similar articles
-
BRD4 Couples NF-κB/RelA with Airway Inflammation and the IRF-RIG-I Amplification Loop in Respiratory Syncytial Virus Infection.J Virol. 2017 Feb 28;91(6):e00007-17. doi: 10.1128/JVI.00007-17. Print 2017 Mar 15. J Virol. 2017. PMID: 28077651 Free PMC article.
-
Retinoic acid-inducible gene I mediates early antiviral response and Toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells.J Virol. 2007 Feb;81(3):1401-11. doi: 10.1128/JVI.01740-06. Epub 2006 Nov 15. J Virol. 2007. PMID: 17108032 Free PMC article.
-
TRAF6 establishes innate immune responses by activating NF-kappaB and IRF7 upon sensing cytosolic viral RNA and DNA.PLoS One. 2009 May 25;4(5):e5674. doi: 10.1371/journal.pone.0005674. PLoS One. 2009. PMID: 19479062 Free PMC article.
-
RIG-I in RNA virus recognition.Virology. 2015 May;479-480:110-21. doi: 10.1016/j.virol.2015.02.017. Epub 2015 Mar 5. Virology. 2015. PMID: 25749629 Free PMC article. Review.
-
The interferon response to intracellular DNA: why so many receptors?Immunobiology. 2013 Nov;218(11):1312-21. doi: 10.1016/j.imbio.2013.07.007. Epub 2013 Jul 29. Immunobiology. 2013. PMID: 23962476 Review.
Cited by
-
Early innate immune response triggered by the human respiratory syncytial virus and its regulation by ubiquitination/deubiquitination processes.J Biomed Sci. 2022 Feb 13;29(1):11. doi: 10.1186/s12929-022-00793-3. J Biomed Sci. 2022. PMID: 35152905 Free PMC article. Review.
-
Therapeutic targets for inflammation-mediated airway remodeling in chronic lung disease.Expert Rev Respir Med. 2018 Nov;12(11):931-939. doi: 10.1080/17476348.2018.1526677. Epub 2018 Oct 3. Expert Rev Respir Med. 2018. PMID: 30241450 Free PMC article. Review.
-
IRF7: role and regulation in immunity and autoimmunity.Front Immunol. 2023 Aug 10;14:1236923. doi: 10.3389/fimmu.2023.1236923. eCollection 2023. Front Immunol. 2023. PMID: 37638030 Free PMC article. Review.
-
Interferon regulatory factor 7 regulates airway epithelial cell responses to human rhinovirus infection.BMC Genomics. 2016 Jan 25;17:76. doi: 10.1186/s12864-016-2405-z. BMC Genomics. 2016. PMID: 26810609 Free PMC article.
-
Pseudorabies Virus Infection Triggers NF-κB Activation via the DNA Damage Response but Actively Inhibits NF-κB-Dependent Gene Expression.J Virol. 2021 Nov 23;95(24):e0166621. doi: 10.1128/JVI.01666-21. Epub 2021 Oct 6. J Virol. 2021. PMID: 34613805 Free PMC article.
References
-
- Glezen WP, Taber LH, Frank AL, Kasel JA. 1986. Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child 140:543–546. - PubMed
-
- Nair H, Nokes DJ, Gessner BD, Dherani M, Madhi SA, Singleton RJ, O'Brien KL, Roca A, Wright PF, Bruce N, Chandran A, Theodoratou E, Sutanto A, Sedyaningsih ER, Ngama M, Munywoki PK, Kartasasmita C, Simoes EA, Rudan I, Weber MW, Campbell H. 2010. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: a systematic review and meta-analysis. Lancet 375:1545–1555. doi:10.1016/S0140-6736(10)60206-1. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
