

pmx: Automated protein structure and topology generation for alchemical perturbations
- PMID: 25487359
- PMCID: PMC4365728
- DOI: 10.1002/jcc.23804
pmx: Automated protein structure and topology generation for alchemical perturbations
Abstract
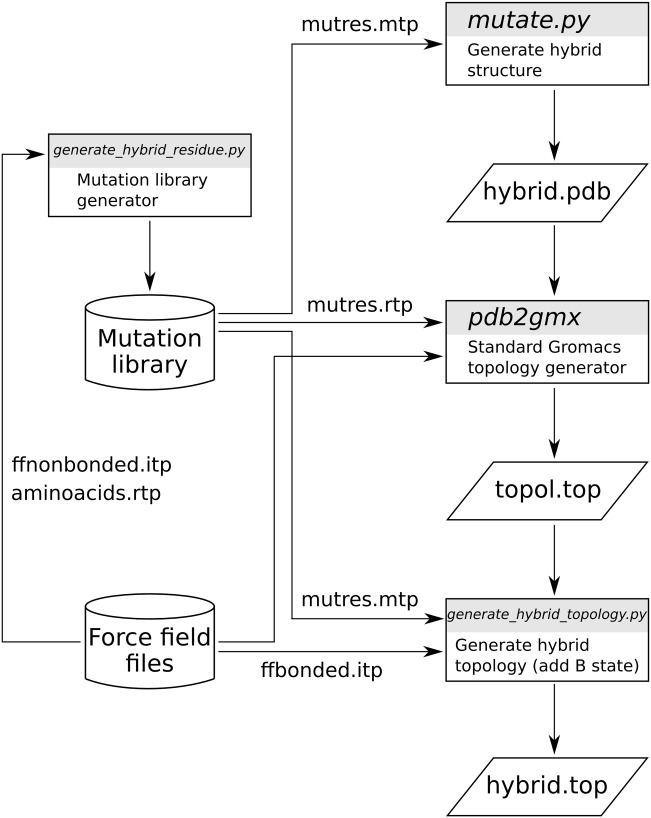
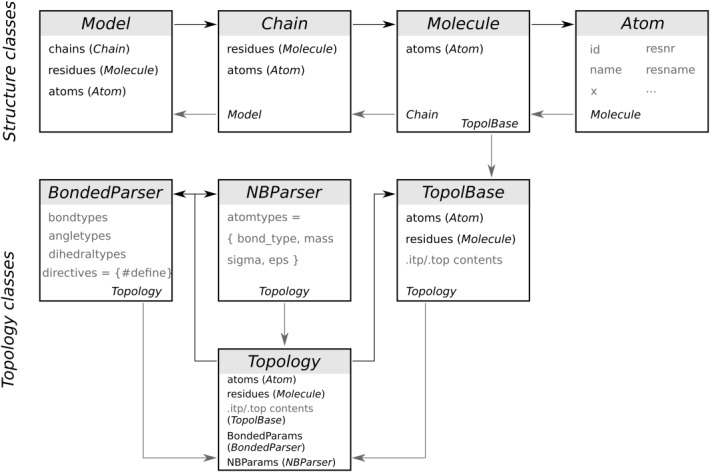
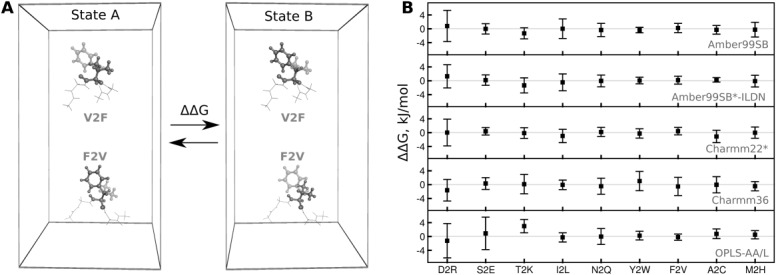
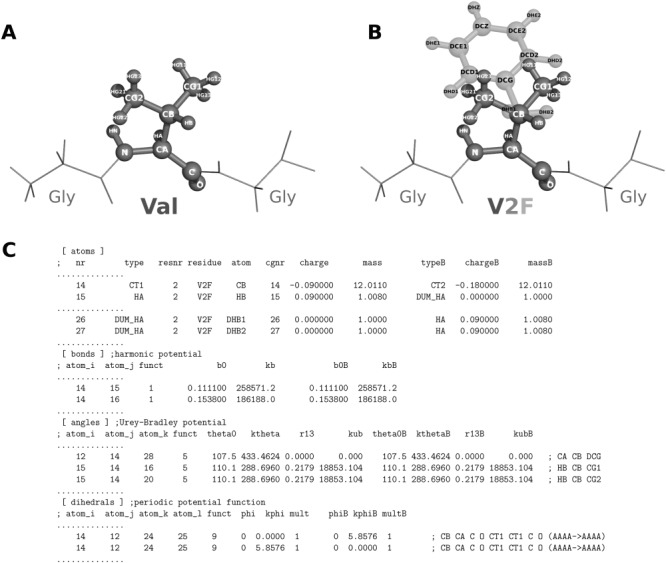
Computational protein design requires methods to accurately estimate free energy changes in protein stability or binding upon an amino acid mutation. From the different approaches available, molecular dynamics-based alchemical free energy calculations are unique in their accuracy and solid theoretical basis. The challenge in using these methods lies in the need to generate hybrid structures and topologies representing two physical states of a system. A custom made hybrid topology may prove useful for a particular mutation of interest, however, a high throughput mutation analysis calls for a more general approach. In this work, we present an automated procedure to generate hybrid structures and topologies for the amino acid mutations in all commonly used force fields. The described software is compatible with the Gromacs simulation package. The mutation libraries are readily supported for five force fields, namely Amber99SB, Amber99SB*-ILDN, OPLS-AA/L, Charmm22*, and Charmm36.
Keywords: alchemy; free energy calculations; molecular dynamics; mutations; thermostability.
© 2014 The Authors Journal of Computational Chemistry Published by Wiley Periodicals, Inc.
Figures
Similar articles
-
Accurate Calculation of Free Energy Changes upon Amino Acid Mutation.Methods Mol Biol. 2019;1851:19-47. doi: 10.1007/978-1-4939-8736-8_2. Methods Mol Biol. 2019. PMID: 30298390
-
pmx Webserver: A User Friendly Interface for Alchemistry.J Chem Inf Model. 2017 Feb 27;57(2):109-114. doi: 10.1021/acs.jcim.6b00498. Epub 2017 Feb 16. J Chem Inf Model. 2017. PMID: 28181802
-
Automatic GROMACS topology generation and comparisons of force fields for solvation free energy calculations.J Phys Chem B. 2015 Jan 22;119(3):810-23. doi: 10.1021/jp505332p. Epub 2014 Nov 7. J Phys Chem B. 2015. PMID: 25343332
-
Comparison of protein force fields for molecular dynamics simulations.Methods Mol Biol. 2008;443:63-88. doi: 10.1007/978-1-59745-177-2_4. Methods Mol Biol. 2008. PMID: 18446282 Review.
-
Advances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins at constant pH.Adv Protein Chem Struct Biol. 2011;85:281-322. doi: 10.1016/B978-0-12-386485-7.00008-9. Adv Protein Chem Struct Biol. 2011. PMID: 21920327 Review.
Cited by
-
Molecular basis for the increased affinity of an RNA recognition motif with re-engineered specificity: A molecular dynamics and enhanced sampling simulations study.PLoS Comput Biol. 2018 Dec 6;14(12):e1006642. doi: 10.1371/journal.pcbi.1006642. eCollection 2018 Dec. PLoS Comput Biol. 2018. PMID: 30521520 Free PMC article.
-
Computing Relative Binding Affinity of Ligands to Receptor: An Effective Hybrid Single-Dual-Topology Free-Energy Perturbation Approach in NAMD.J Chem Inf Model. 2019 Sep 23;59(9):3794-3802. doi: 10.1021/acs.jcim.9b00362. Epub 2019 Aug 27. J Chem Inf Model. 2019. PMID: 31411473 Free PMC article.
-
Evaluating inositol phospholipid interactions with inward rectifier potassium channels and characterising their role in disease.Commun Chem. 2020 Oct 30;3(1):147. doi: 10.1038/s42004-020-00391-0. Commun Chem. 2020. PMID: 36703430 Free PMC article.
-
Performance evaluation of molecular docking and free energy calculations protocols using the D3R Grand Challenge 4 dataset.J Comput Aided Mol Des. 2019 Dec;33(12):1031-1043. doi: 10.1007/s10822-019-00232-w. Epub 2019 Nov 1. J Comput Aided Mol Des. 2019. PMID: 31677003
-
Commensal bacteria stimulate antitumor responses via T cell cross-reactivity.JCI Insight. 2020 Apr 23;5(8):e135597. doi: 10.1172/jci.insight.135597. JCI Insight. 2020. PMID: 32324171 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
