

m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells
- PMID: 25456834
- PMCID: PMC4278749
- DOI: 10.1016/j.stem.2014.09.019
m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells
Abstract
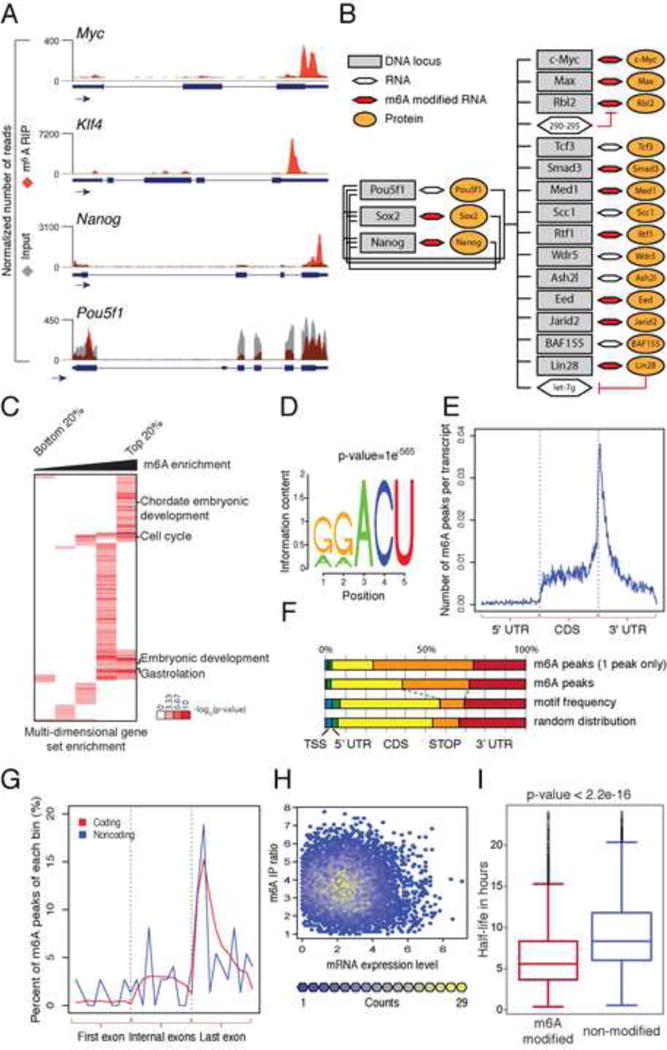
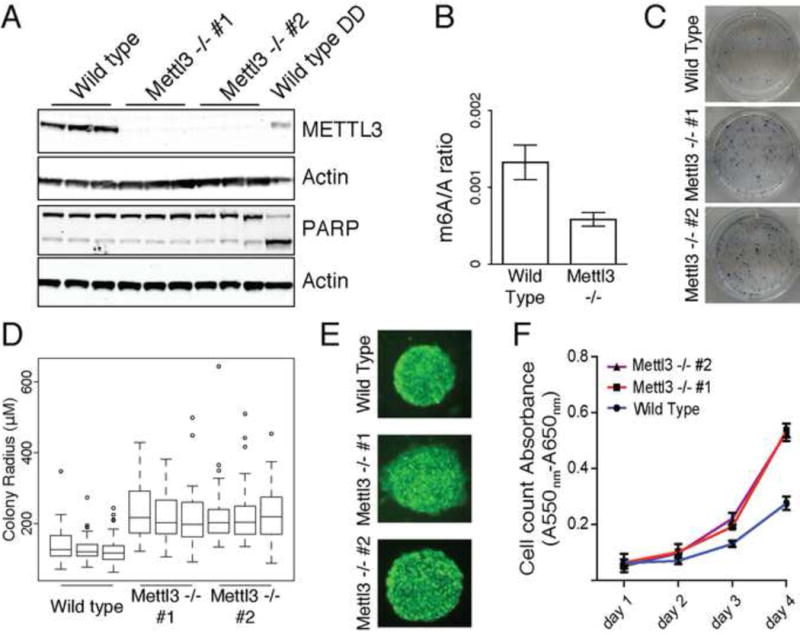
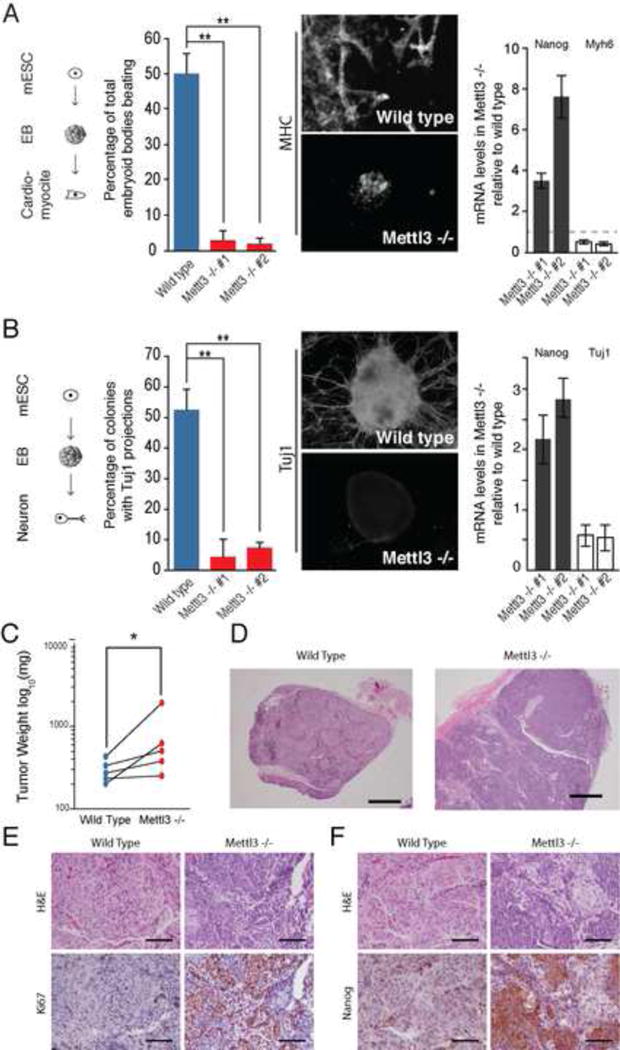
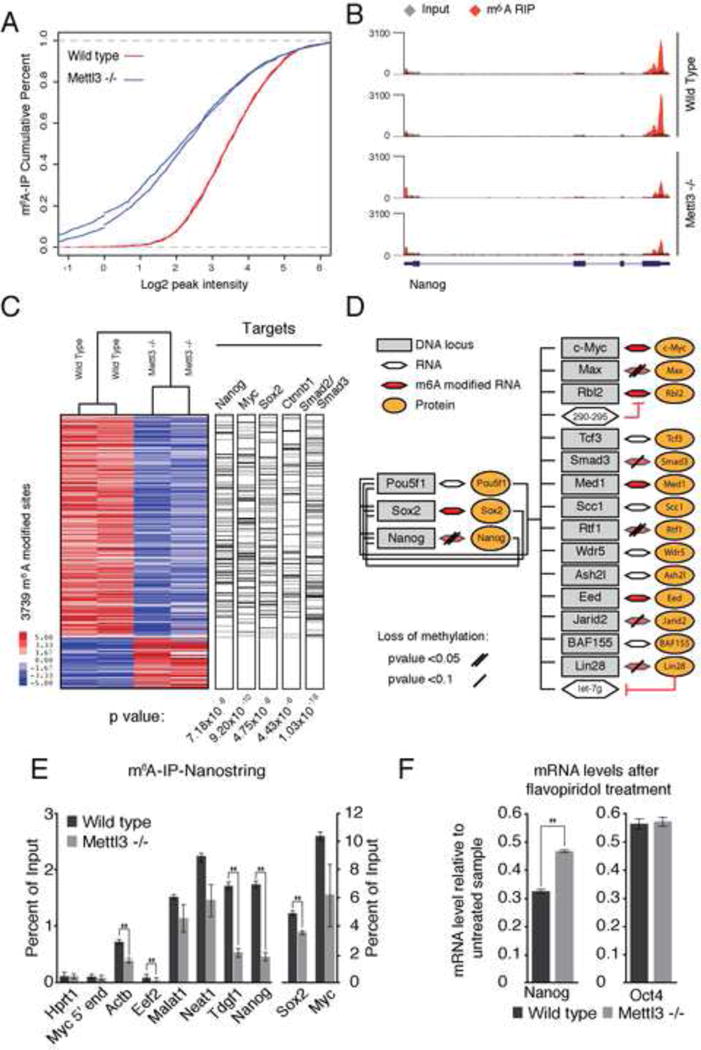
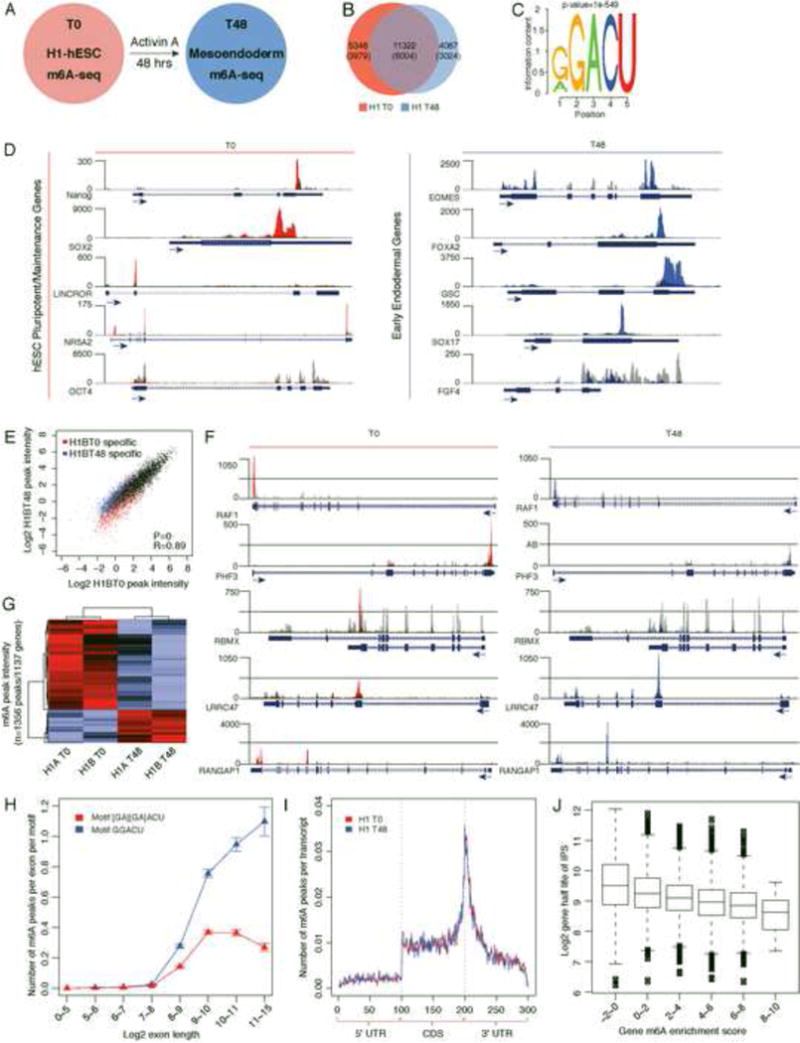
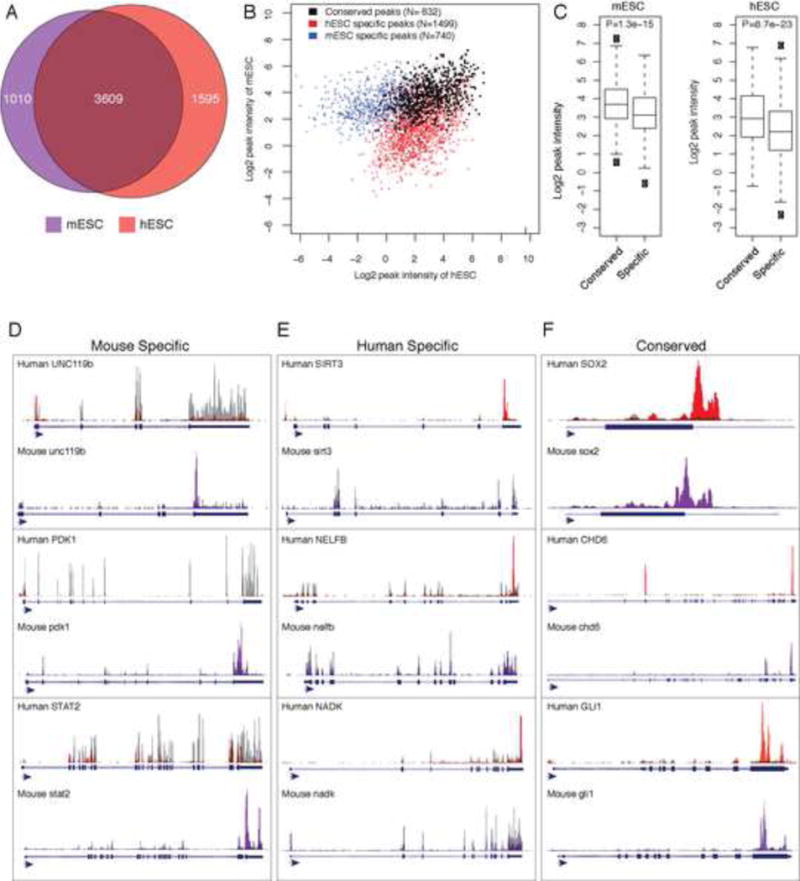
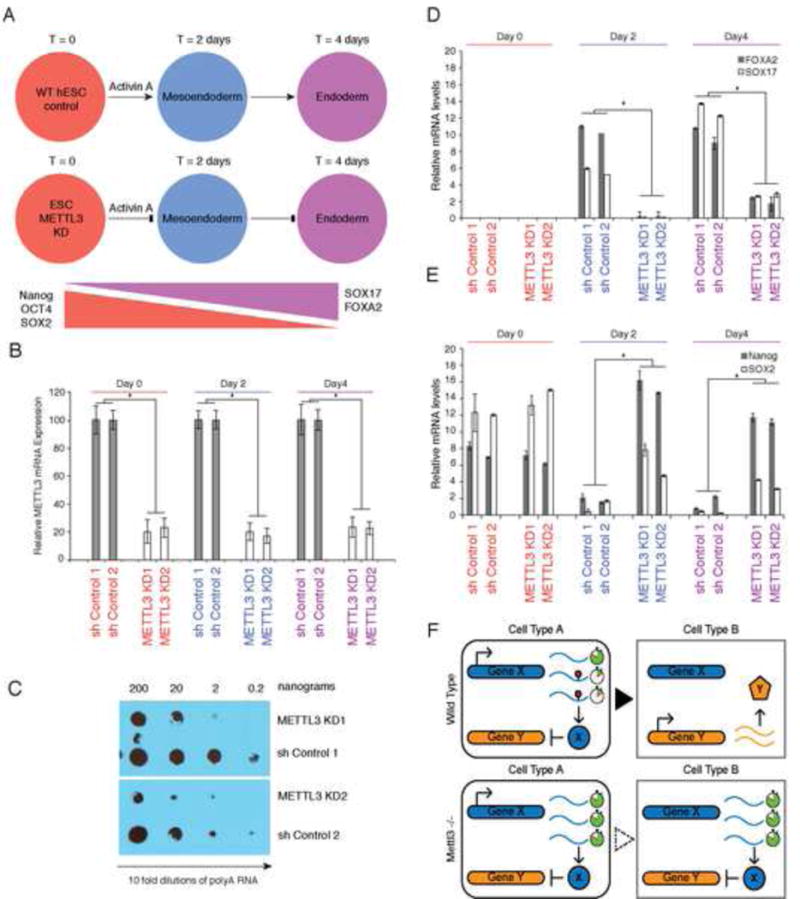
N6-methyl-adenosine (m(6)A) is the most abundant modification on messenger RNAs and is linked to human diseases, but its functions in mammalian development are poorly understood. Here we reveal the evolutionary conservation and function of m(6)A by mapping the m(6)A methylome in mouse and human embryonic stem cells. Thousands of messenger and long noncoding RNAs show conserved m(6)A modification, including transcripts encoding core pluripotency transcription factors. m(6)A is enriched over 3' untranslated regions at defined sequence motifs and marks unstable transcripts, including transcripts turned over upon differentiation. Genetic inactivation or depletion of mouse and human Mettl3, one of the m(6)A methylases, led to m(6)A erasure on select target genes, prolonged Nanog expression upon differentiation, and impaired ESC exit from self-renewal toward differentiation into several lineages in vitro and in vivo. Thus, m(6)A is a mark of transcriptome flexibility required for stem cells to differentiate to specific lineages.
Copyright © 2014 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Stem cell RNA epigenetics: m(6)arking your territory.Cell Stem Cell. 2014 Dec 4;15(6):669-70. doi: 10.1016/j.stem.2014.11.011. Cell Stem Cell. 2014. PMID: 25479740
Similar articles
-
Stem cell RNA epigenetics: m(6)arking your territory.Cell Stem Cell. 2014 Dec 4;15(6):669-70. doi: 10.1016/j.stem.2014.11.011. Cell Stem Cell. 2014. PMID: 25479740
-
N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells.Nat Cell Biol. 2014 Feb;16(2):191-8. doi: 10.1038/ncb2902. Epub 2014 Jan 7. Nat Cell Biol. 2014. PMID: 24394384 Free PMC article.
-
Simultaneous overexpression of Oct4 and Nanog abrogates terminal myogenesis.Am J Physiol Cell Physiol. 2009 Jul;297(1):C43-54. doi: 10.1152/ajpcell.00468.2008. Epub 2009 Apr 29. Am J Physiol Cell Physiol. 2009. PMID: 19403798
-
Nanog and transcriptional networks in embryonic stem cell pluripotency.Cell Res. 2007 Jan;17(1):42-9. doi: 10.1038/sj.cr.7310125. Cell Res. 2007. PMID: 17211451 Review.
-
METTLing in Stem Cell and Cancer Biology.Stem Cell Rev Rep. 2023 Jan;19(1):76-91. doi: 10.1007/s12015-022-10444-7. Epub 2022 Sep 12. Stem Cell Rev Rep. 2023. PMID: 36094754 Free PMC article. Review.
Cited by
-
Global Co-regulatory Cross Talk Between m6A and m5C RNA Methylation Systems Coordinate Cellular Responses and Brain Disease Pathways.Mol Neurobiol. 2024 Nov 5. doi: 10.1007/s12035-024-04555-0. Online ahead of print. Mol Neurobiol. 2024. PMID: 39499421
-
New insights into the epitranscriptomic control of pluripotent stem cell fate.Exp Mol Med. 2022 Oct;54(10):1643-1651. doi: 10.1038/s12276-022-00824-x. Epub 2022 Oct 21. Exp Mol Med. 2022. PMID: 36266446 Free PMC article. Review.
-
Epigenetic mechanisms in neurogenesis.Nat Rev Neurosci. 2016 Sep;17(9):537-49. doi: 10.1038/nrn.2016.70. Epub 2016 Jun 23. Nat Rev Neurosci. 2016. PMID: 27334043 Free PMC article. Review.
-
Resetting the epigenome: Methylation dynamics in cancer stem cells.Front Cell Dev Biol. 2022 Sep 26;10:909424. doi: 10.3389/fcell.2022.909424. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36225315 Free PMC article. Review.
-
Biphasic Liquid Microjunction Extraction for Profiling Neuronal RNA Modifications by Liquid Chromatography-Tandem Mass Spectrometry.Anal Chem. 2020 Sep 15;92(18):12647-12655. doi: 10.1021/acs.analchem.0c02830. Epub 2020 Aug 27. Anal Chem. 2020. PMID: 32786436 Free PMC article.
References
-
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
