

Self-assembled FUS binds active chromatin and regulates gene transcription
- PMID: 25453086
- PMCID: PMC4273402
- DOI: 10.1073/pnas.1414004111
Self-assembled FUS binds active chromatin and regulates gene transcription
Abstract
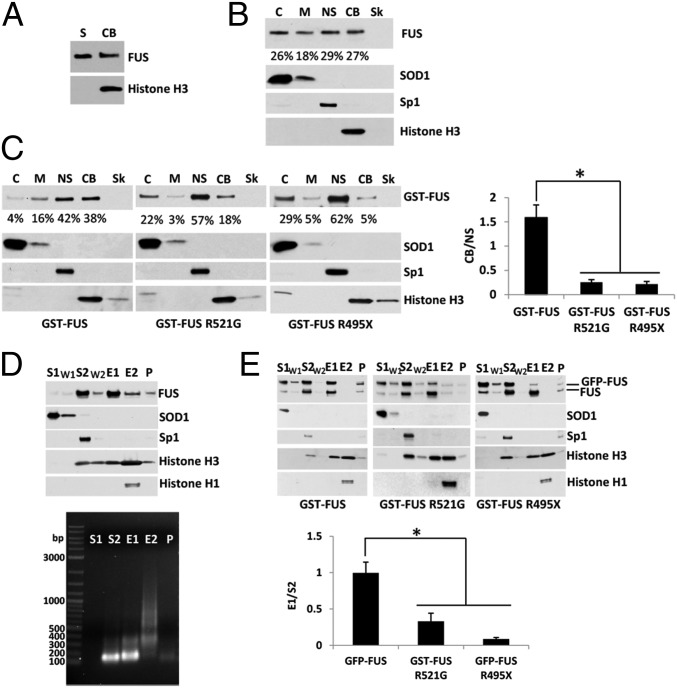
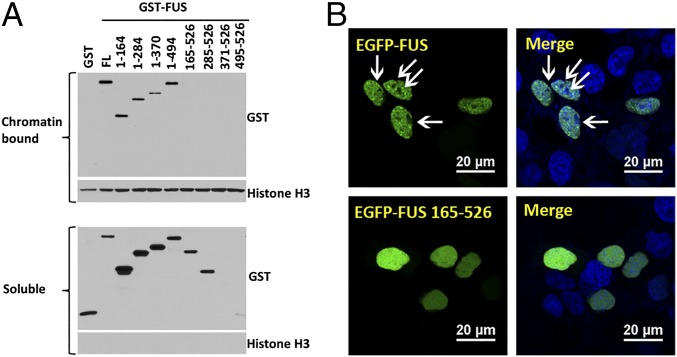
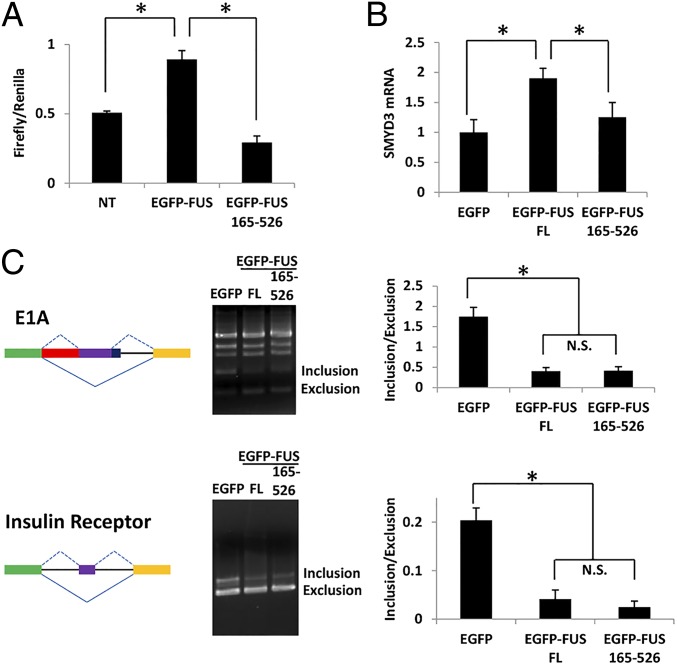
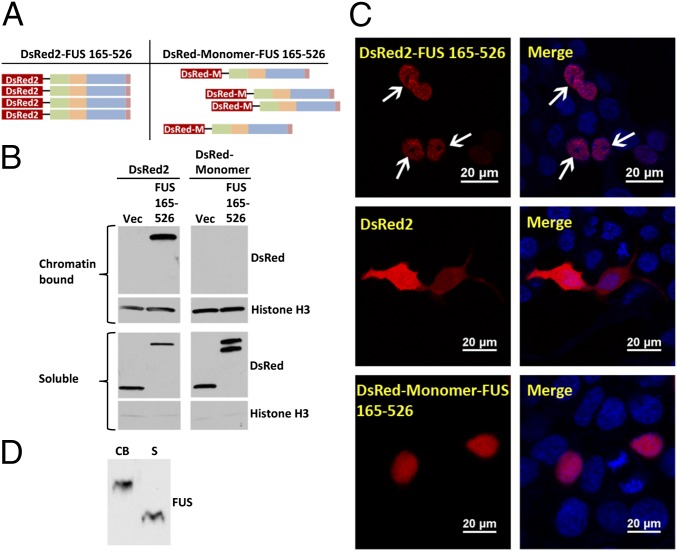
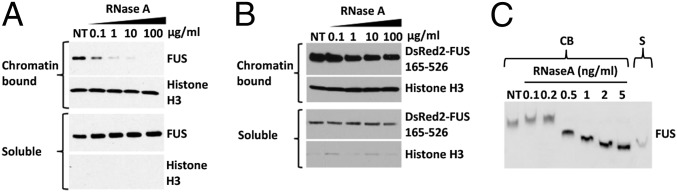
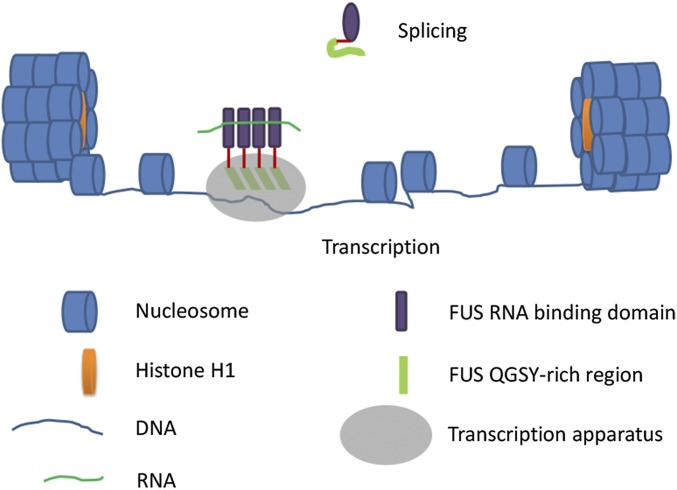
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease. Fused in sarcoma (FUS) is a DNA/RNA binding protein and mutations in FUS cause a subset of familial ALS. Most ALS mutations are clustered in the C-terminal nuclear localization sequence of FUS and consequently lead to the accumulation of protein inclusions in the cytoplasm. It remains debatable whether loss of FUS normal function in the nucleus or gain of toxic function in the cytoplasm plays a more critical role in the ALS etiology. Moreover, the physiological function of FUS in the nucleus remains to be fully understood. In this study, we found that a significant portion of nuclear FUS was bound to active chromatin and that the ALS mutations dramatically decreased FUS chromatin binding ability. Functionally, the chromatin binding is required for FUS transcription activation, but not for alternative splicing regulation. The N-terminal QGSY (glutamine-glycine-serine-tyrosine)-rich region (amino acids 1-164) mediates FUS self-assembly in the nucleus of mammalian cells and the self-assembly is essential for its chromatin binding and transcription activation. In addition, RNA binding is also required for FUS self-assembly and chromatin binding. Together, our results suggest a functional assembly of FUS in the nucleus under physiological conditions, which is different from the cytoplasmic inclusions. The ALS mutations can cause loss of function in the nucleus by disrupting this assembly and chromatin binding.
Keywords: amyotrophic lateral sclerosis; chromatin binding; fused in sarcoma; self-assembly; transcription.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
FUS interacts with nuclear matrix-associated protein SAFB1 as well as Matrin3 to regulate splicing and ligand-mediated transcription.Sci Rep. 2016 Oct 12;6:35195. doi: 10.1038/srep35195. Sci Rep. 2016. PMID: 27731383 Free PMC article.
-
Subcellular localization and RNAs determine FUS architecture in different cellular compartments.Hum Mol Genet. 2015 Sep 15;24(18):5174-83. doi: 10.1093/hmg/ddv239. Epub 2015 Jun 29. Hum Mol Genet. 2015. PMID: 26123490 Free PMC article.
-
RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations.Hum Mol Genet. 2013 Mar 15;22(6):1193-205. doi: 10.1093/hmg/dds526. Epub 2012 Dec 20. Hum Mol Genet. 2013. PMID: 23257289 Free PMC article.
-
Fused in sarcoma (FUS): an oncogene goes awry in neurodegeneration.Mol Cell Neurosci. 2013 Sep;56:475-86. doi: 10.1016/j.mcn.2013.03.006. Epub 2013 Apr 2. Mol Cell Neurosci. 2013. PMID: 23557964 Review.
-
FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis.Brain Res. 2012 Jun 26;1462:40-3. doi: 10.1016/j.brainres.2011.12.010. Epub 2011 Dec 13. Brain Res. 2012. PMID: 22261247 Review.
Cited by
-
A phase-separated nuclear GBPL circuit controls immunity in plants.Nature. 2021 Jun;594(7863):424-429. doi: 10.1038/s41586-021-03572-6. Epub 2021 May 26. Nature. 2021. PMID: 34040255 Free PMC article.
-
hnRNPs: roles in neurodevelopment and implication for brain disorders.Front Mol Neurosci. 2024 Jul 17;17:1411639. doi: 10.3389/fnmol.2024.1411639. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39086926 Free PMC article. Review.
-
Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations.Mol Cell. 2020 Jan 2;77(1):82-94.e4. doi: 10.1016/j.molcel.2019.09.022. Epub 2019 Oct 17. Mol Cell. 2020. PMID: 31630970 Free PMC article.
-
Regulation of zebrafish dorsoventral patterning by phase separation of RNA-binding protein Rbm14.Cell Discov. 2019 Jul 23;5:37. doi: 10.1038/s41421-019-0106-x. eCollection 2019. Cell Discov. 2019. PMID: 31636951 Free PMC article.
-
Dynamic cycling of t-SNARE acylation regulates platelet exocytosis.J Biol Chem. 2018 Mar 9;293(10):3593-3606. doi: 10.1074/jbc.RA117.000140. Epub 2018 Jan 19. J Biol Chem. 2018. PMID: 29352103 Free PMC article.
References
-
- Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
