

Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP
- PMID: 25406263
- PMCID: PMC4297778
- DOI: 10.1152/ajpendo.00344.2014
Somatostatin and insulin mediate glucose-inhibited glucagon secretion in the pancreatic α-cell by lowering cAMP
Abstract
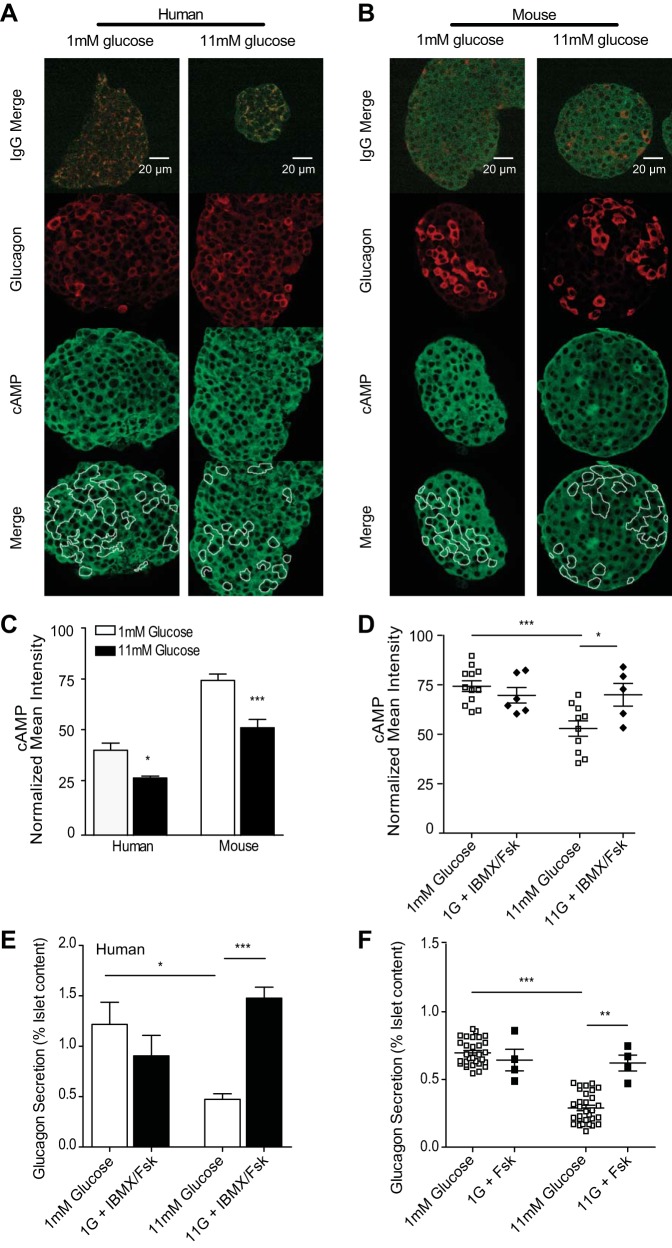
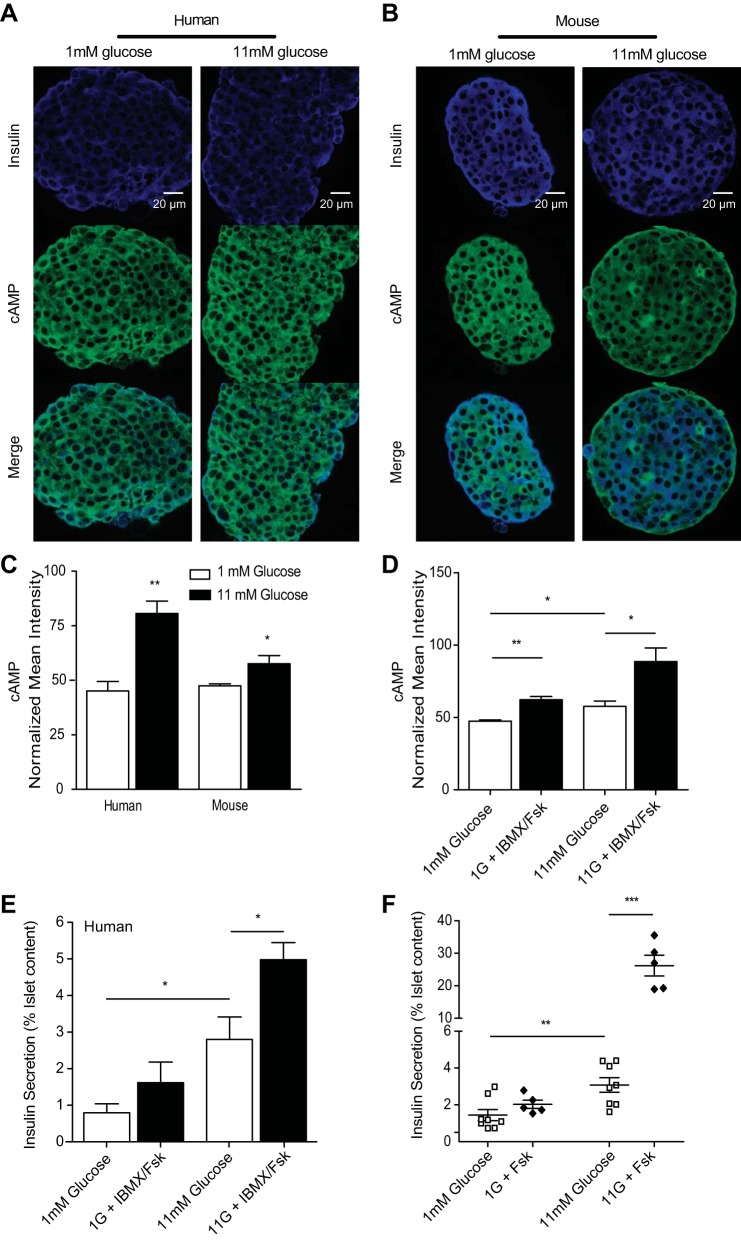
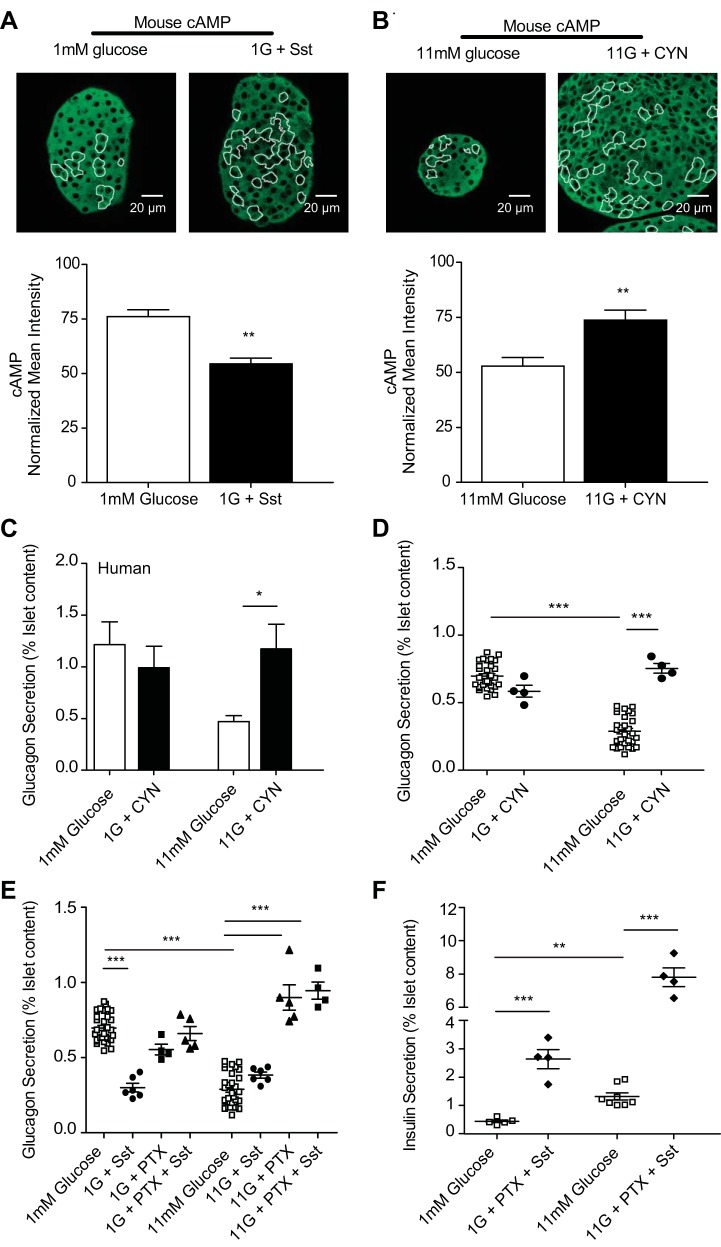
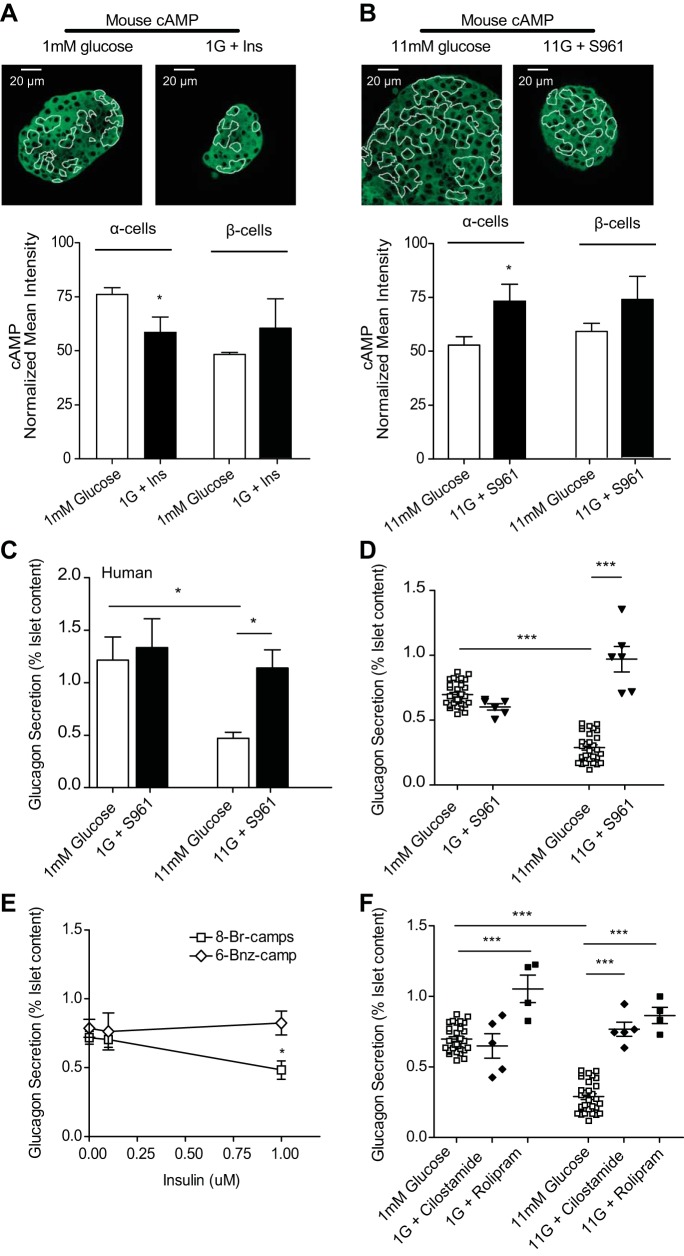
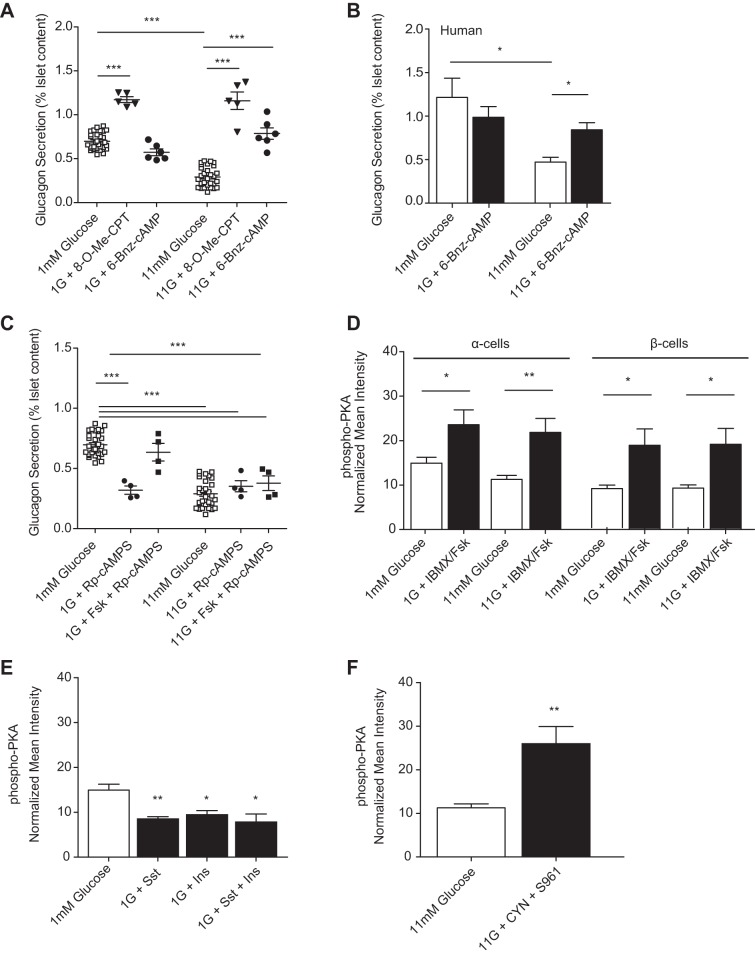
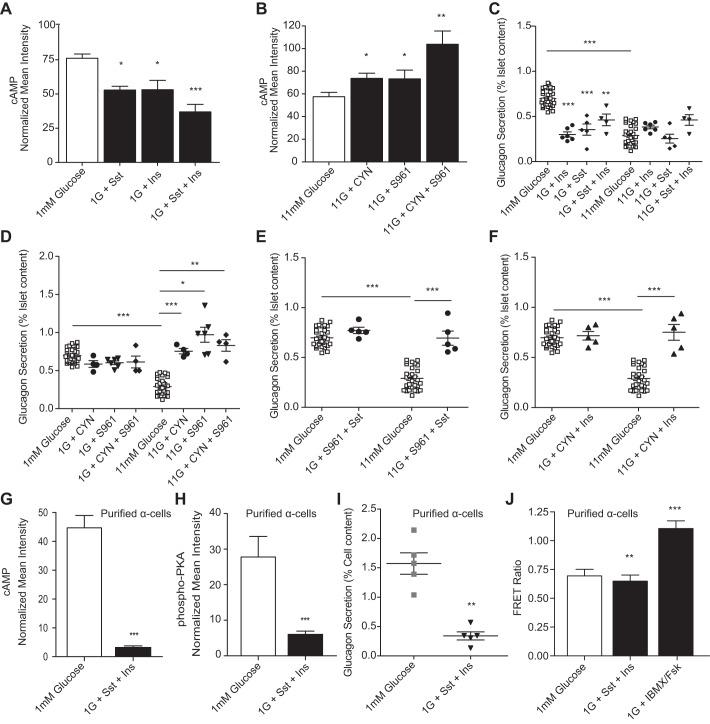
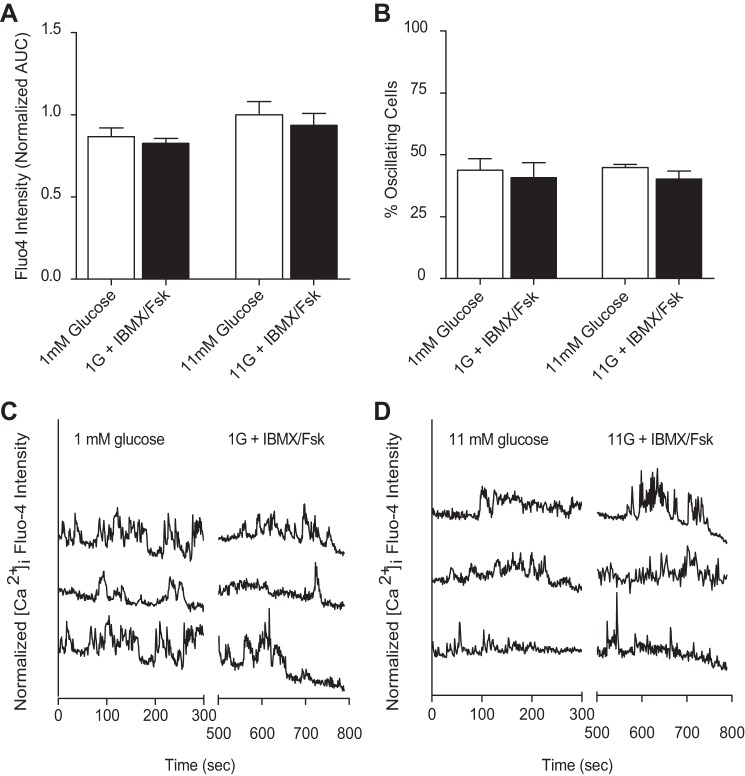
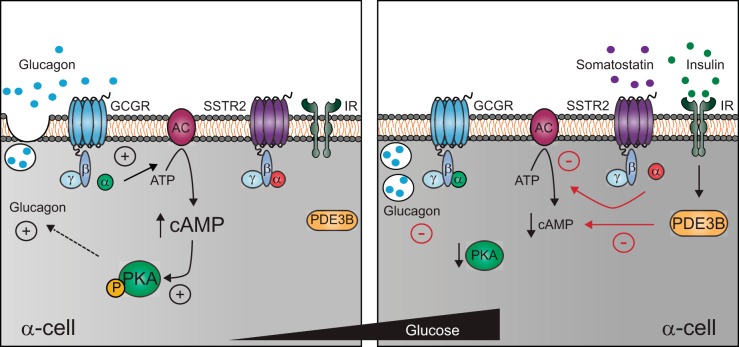
The dysregulation of glucose-inhibited glucagon secretion from the pancreatic islet α-cell is a critical component of diabetes pathology and metabolic disease. We show a previously uncharacterized [Ca(2+)]i-independent mechanism of glucagon suppression in human and murine pancreatic islets whereby cAMP and PKA signaling are decreased. This decrease is driven by the combination of somatostatin, which inhibits adenylyl cyclase production of cAMP via the Gαi subunit of the SSTR2, and insulin, which acts via its receptor to activate phosphodiesterase 3B and degrade cytosolic cAMP. Our data indicate that both somatostatin and insulin signaling are required to suppress cAMP/PKA and glucagon secretion from both human and murine α-cells, and the combination of these two signaling mechanisms is sufficient to reduce glucagon secretion from isolated α-cells as well as islets. Thus, we conclude that somatostatin and insulin together are critical paracrine mediators of glucose-inhibited glucagon secretion and function by lowering cAMP/PKA signaling with increasing glucose.
Keywords: cyclic AMP; glucagon; insulin; pancreatic islets; somatostatin.
Copyright © 2015 the American Physiological Society.
Figures
Similar articles
-
Glucose controls glucagon secretion by directly modulating cAMP in alpha cells.Diabetologia. 2019 Jul;62(7):1212-1224. doi: 10.1007/s00125-019-4857-6. Epub 2019 Apr 5. Diabetologia. 2019. PMID: 30953108 Free PMC article.
-
Free fatty acid receptor 4 inhibitory signaling in delta cells regulates islet hormone secretion in mice.Mol Metab. 2021 Mar;45:101166. doi: 10.1016/j.molmet.2021.101166. Epub 2021 Jan 20. Mol Metab. 2021. PMID: 33484949 Free PMC article.
-
Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice.Endocrinology. 2000 Jan;141(1):111-7. doi: 10.1210/endo.141.1.7263. Endocrinology. 2000. PMID: 10614629
-
cAMP signalling in insulin and glucagon secretion.Diabetes Obes Metab. 2017 Sep;19 Suppl 1:42-53. doi: 10.1111/dom.12993. Diabetes Obes Metab. 2017. PMID: 28466587 Review.
-
The somatostatin receptor in human pancreatic β-cells.Vitam Horm. 2014;95:165-93. doi: 10.1016/B978-0-12-800174-5.00007-7. Vitam Horm. 2014. PMID: 24559918 Review.
Cited by
-
Cell Autonomous Dysfunction and Insulin Resistance in Pancreatic α Cells.Int J Mol Sci. 2019 Jul 28;20(15):3699. doi: 10.3390/ijms20153699. Int J Mol Sci. 2019. PMID: 31357734 Free PMC article. Review.
-
Leucine Suppresses α-Cell cAMP and Glucagon Secretion via a Combination of Cell-Intrinsic and Islet Paracrine Signaling.Diabetes. 2024 Sep 1;73(9):1426-1439. doi: 10.2337/db23-1013. Diabetes. 2024. PMID: 38870025
-
Mechanism of insulin resistance in obesity: a role of ATP.Front Med. 2021 Jun;15(3):372-382. doi: 10.1007/s11684-021-0862-5. Epub 2021 May 28. Front Med. 2021. PMID: 34047935 Review.
-
Growth Hormone-Releasing Hormone in Diabetes.Front Endocrinol (Lausanne). 2016 Oct 10;7:129. doi: 10.3389/fendo.2016.00129. eCollection 2016. Front Endocrinol (Lausanne). 2016. PMID: 27777568 Free PMC article. Review.
-
RhoA as a Signaling Hub Controlling Glucagon Secretion From Pancreatic α-Cells.Diabetes. 2022 Nov 1;71(11):2384-2394. doi: 10.2337/db21-1010. Diabetes. 2022. PMID: 35904939 Free PMC article.
References
-
- Ahmad F, Lindh R, Tang Y, Ruishalme I, Ost A, Sahachartsiri B, Strålfors P, Degerman E, Manganiello VC. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the beta3-adrenergic receptor agonist Cl316243: effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem J 424: 399–410, 2009. - PMC - PubMed
-
- Beebe SJ, Redmon JB, Blackmore PF, Corbin JD. Discriminative insulin antagonism of stimulatory effects of various cAMP analogs on adipocyte lipolysis and hepatocyte glycogenolysis. J Biol Chem 260: 15781–15788, 1985. - PubMed
-
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520, 2006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous