

Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy
- PMID: 25404307
- PMCID: PMC4260547
- DOI: 10.1073/pnas.1415589111
Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy
Abstract
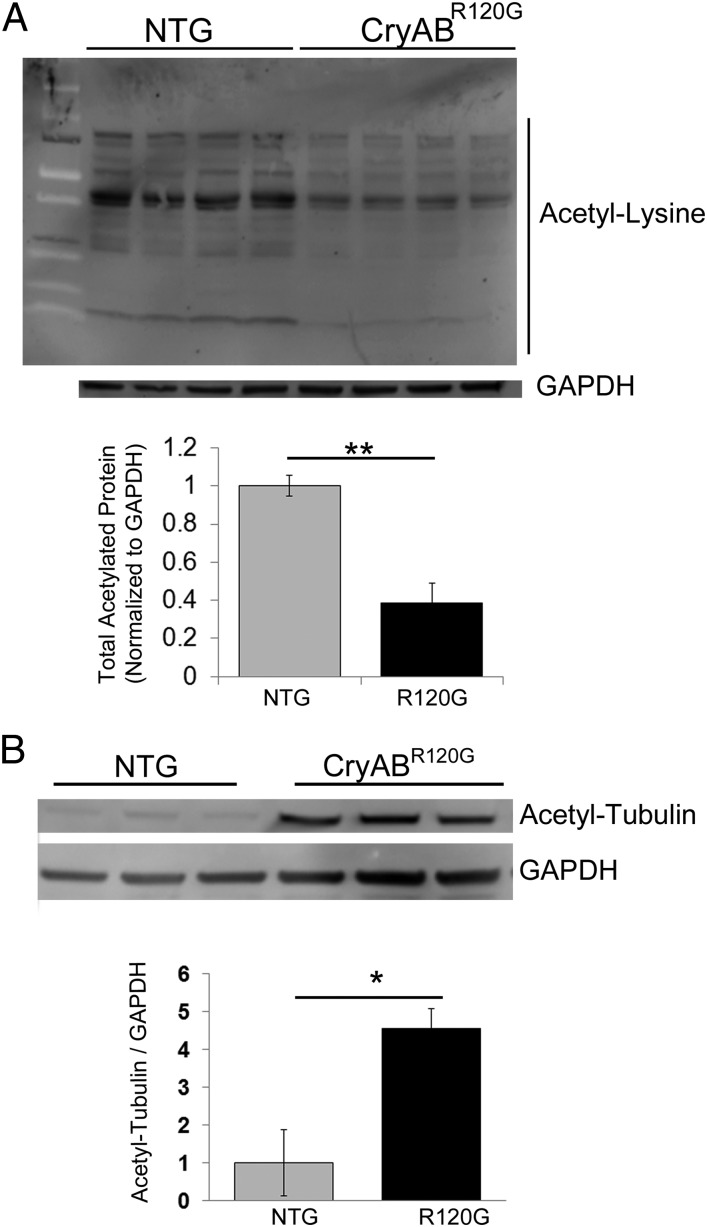
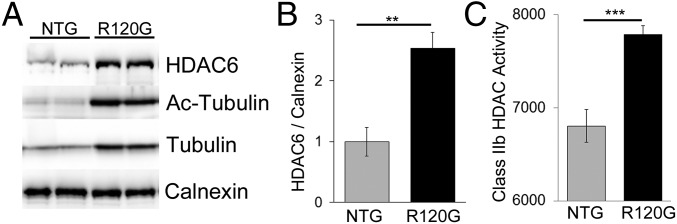
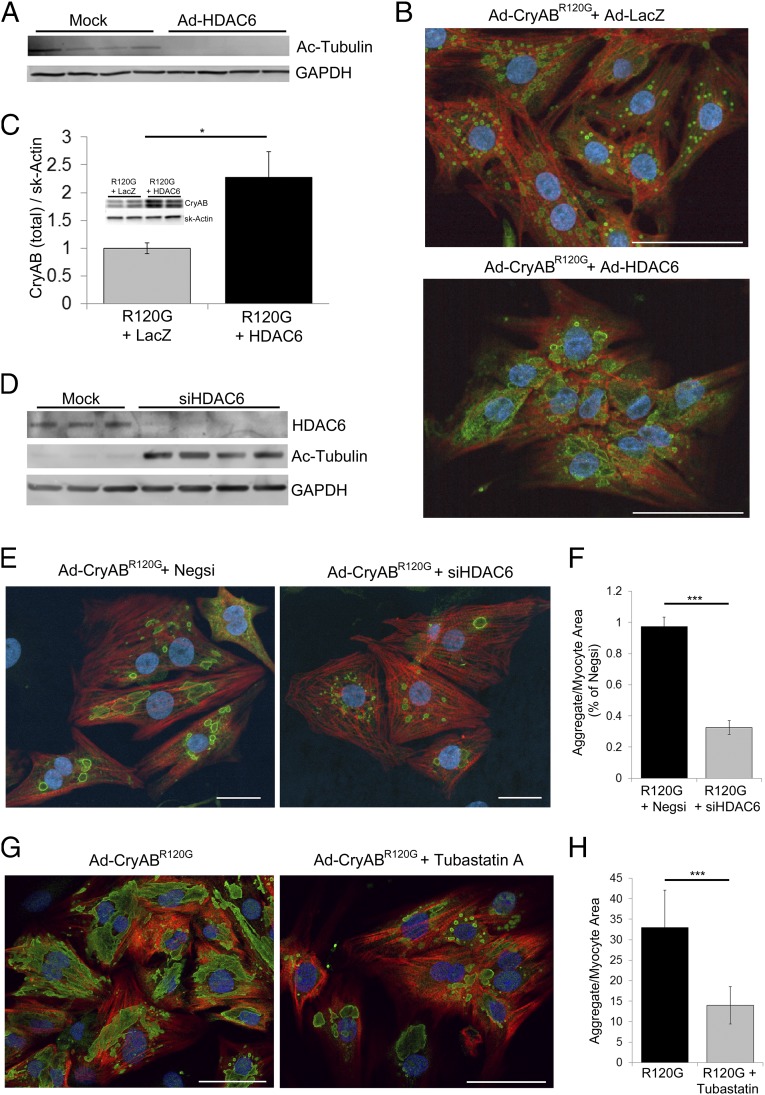
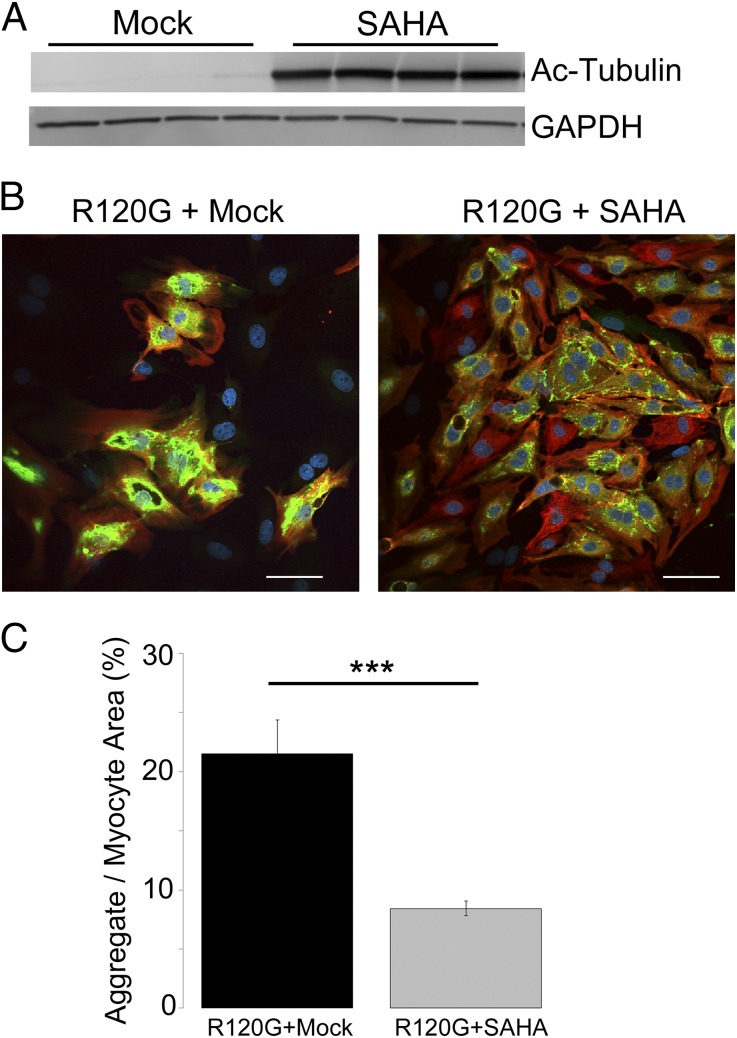
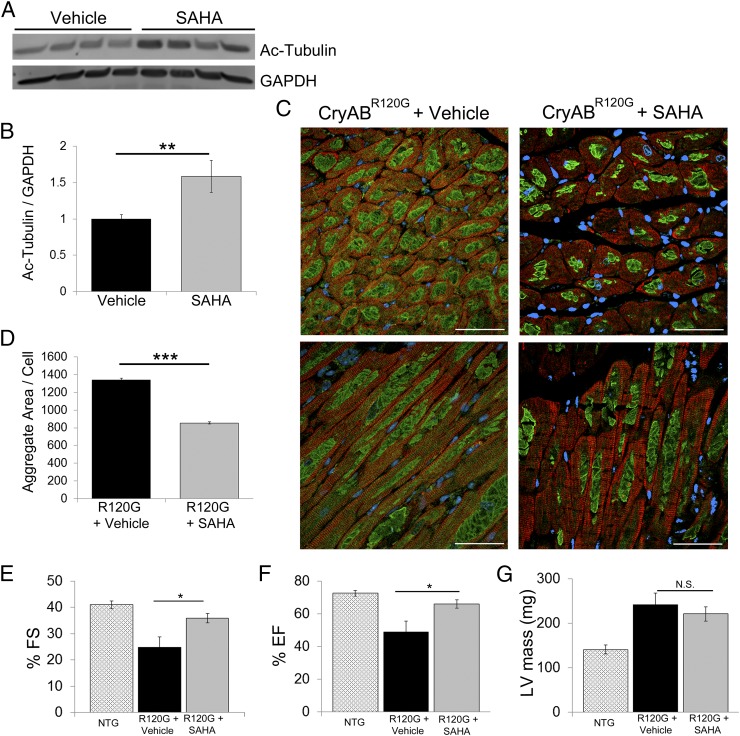
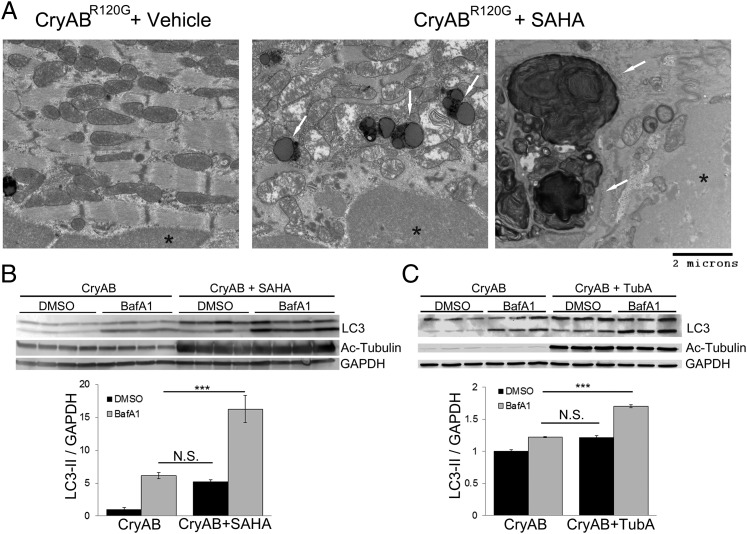
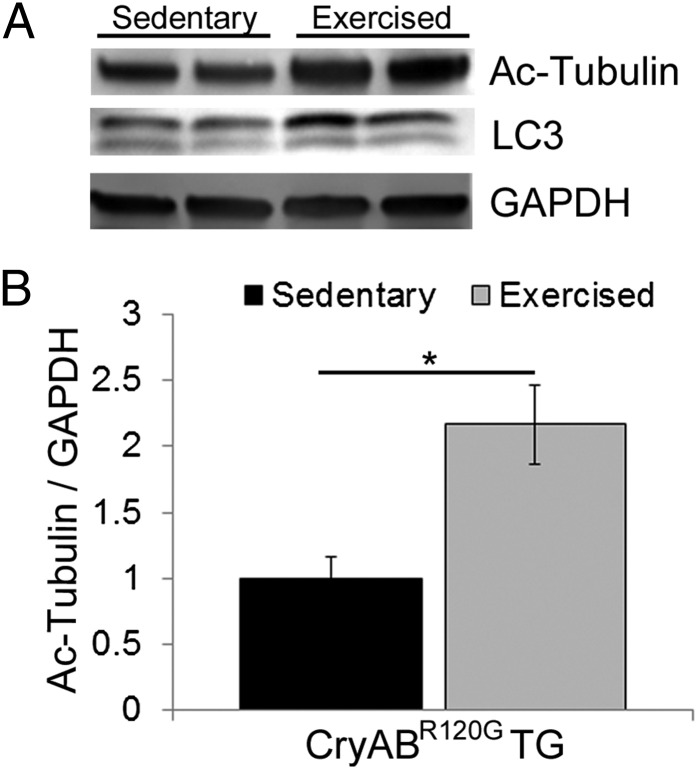
Proteinopathy causes cardiac disease, remodeling, and heart failure but the pathological mechanisms remain obscure. Mutated αB-crystallin (CryAB(R120G)), when expressed only in cardiomyocytes in transgenic (TG) mice, causes desmin-related cardiomyopathy, a protein conformational disorder. The disease is characterized by the accumulation of toxic misfolded protein species that present as perinuclear aggregates known as aggresomes. Previously, we have used the CryAB(R120G) model to determine the underlying processes that result in these pathologic accumulations and to explore potential therapeutic windows that might be used to decrease proteotoxicity. We noted that total ventricular protein is hypoacetylated while hyperacetylation of α-tubulin, a substrate of histone deacetylase 6 (HDAC6) occurs. HDAC6 has critical roles in protein trafficking and autophagy, but its function in the heart is obscure. Here, we test the hypothesis that tubulin acetylation is an adaptive process in cardiomyocytes. By modulating HDAC6 levels and/or activity genetically and pharmacologically, we determined the effects of tubulin acetylation on aggregate formation in CryAB(R120G) cardiomyocytes. Increasing HDAC6 accelerated aggregate formation, whereas siRNA-mediated knockdown or pharmacological inhibition ameliorated the process. HDAC inhibition in vivo induced tubulin hyperacetylation in CryAB(R120G) TG hearts, which prevented aggregate formation and significantly improved cardiac function. HDAC6 inhibition also increased autophagic flux in cardiomyocytes, and increased autophagy in the diseased heart correlated with increased tubulin acetylation, suggesting that autophagy induction might underlie the observed cardioprotection. Taken together, our data suggest a mechanistic link between tubulin hyperacetylation and autophagy induction and points to HDAC6 as a viable therapeutic target in cardiovascular disease.
Keywords: HDAC6; alphaB-crystallin; autophagy; heart; proteotoxicity.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes.Circ Res. 2011 Jul 8;109(2):151-60. doi: 10.1161/CIRCRESAHA.110.237339. Epub 2011 May 26. Circ Res. 2011. PMID: 21617129 Free PMC article.
-
Ube2v1 Positively Regulates Protein Aggregation by Modulating Ubiquitin Proteasome System Performance Partially Through K63 Ubiquitination.Circ Res. 2020 Mar 27;126(7):907-922. doi: 10.1161/CIRCRESAHA.119.316444. Epub 2020 Feb 21. Circ Res. 2020. PMID: 32081062 Free PMC article.
-
COP9 signalosome controls the degradation of cytosolic misfolded proteins and protects against cardiac proteotoxicity.Circ Res. 2015 Nov 6;117(11):956-66. doi: 10.1161/CIRCRESAHA.115.306783. Epub 2015 Sep 17. Circ Res. 2015. PMID: 26383969 Free PMC article.
-
Molecular mechanisms of α-crystallinopathy and its therapeutic strategy.Biol Pharm Bull. 2011;34(11):1653-8. doi: 10.1248/bpb.34.1653. Biol Pharm Bull. 2011. PMID: 22040875 Review.
-
HDAC6 α-tubulin deacetylase: a potential therapeutic target in neurodegenerative diseases.J Neurol Sci. 2011 May 15;304(1-2):1-8. doi: 10.1016/j.jns.2011.02.017. Epub 2011 Mar 5. J Neurol Sci. 2011. PMID: 21377170 Review.
Cited by
-
Histone deacetylase 6 in cancer.J Hematol Oncol. 2018 Sep 3;11(1):111. doi: 10.1186/s13045-018-0654-9. J Hematol Oncol. 2018. PMID: 30176876 Free PMC article. Review.
-
Targeting mitochondria for cardiovascular disorders: therapeutic potential and obstacles.Nat Rev Cardiol. 2019 Jan;16(1):33-55. doi: 10.1038/s41569-018-0074-0. Nat Rev Cardiol. 2019. PMID: 30177752 Free PMC article. Review.
-
Tubulin Post-Translational Modifications: The Elusive Roles of Acetylation.Biology (Basel). 2023 Apr 6;12(4):561. doi: 10.3390/biology12040561. Biology (Basel). 2023. PMID: 37106761 Free PMC article. Review.
-
A Molecular Perspective on Sirtuin Activity.Int J Mol Sci. 2020 Nov 15;21(22):8609. doi: 10.3390/ijms21228609. Int J Mol Sci. 2020. PMID: 33203121 Free PMC article. Review.
-
Posttranslational modifications of α-tubulin in alzheimer disease.Transl Neurodegener. 2015 May 15;4:9. doi: 10.1186/s40035-015-0030-4. eCollection 2015. Transl Neurodegener. 2015. PMID: 26029362 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
- F32 HL112558/HL/NHLBI NIH HHS/United States
- R01 HL116848/HL/NHLBI NIH HHS/United States
- P01HL69779/HL/NHLBI NIH HHS/United States
- R01HL05924/HL/NHLBI NIH HHS/United States
- T32 HL007171/HL/NHLBI NIH HHS/United States
- P01 HL069779/HL/NHLBI NIH HHS/United States
- P01 HL059408/HL/NHLBI NIH HHS/United States
- HL116848/HL/NHLBI NIH HHS/United States
- R21 AG043822/AG/NIA NIH HHS/United States
- R011062927/PHS HHS/United States
- AG043822/AG/NIA NIH HHS/United States
- T32 HL007382/HL/NHLBI NIH HHS/United States
- F32 HL124893/HL/NHLBI NIH HHS/United States
- P01HL059408/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
