

Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) through a pathway involving PPARγ coactivator 1α (PGC1α)
- PMID: 25389293
- PMCID: PMC4294522
- DOI: 10.1074/jbc.M114.589325
Relaxin activates peroxisome proliferator-activated receptor γ (PPARγ) through a pathway involving PPARγ coactivator 1α (PGC1α)
Abstract
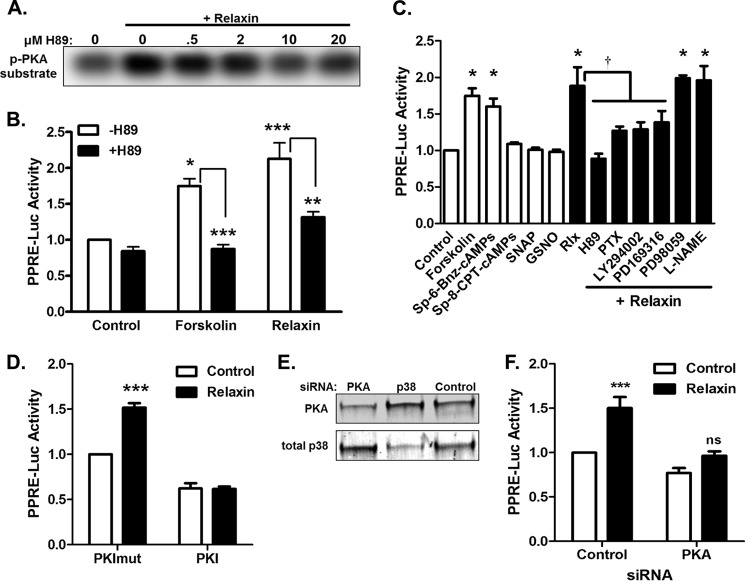
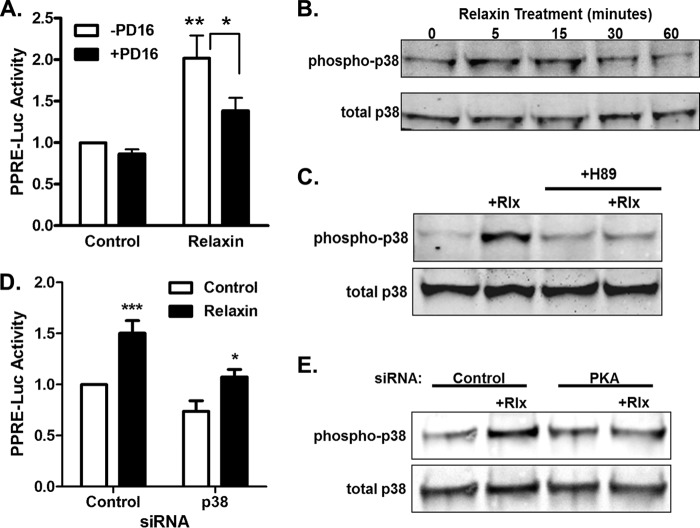
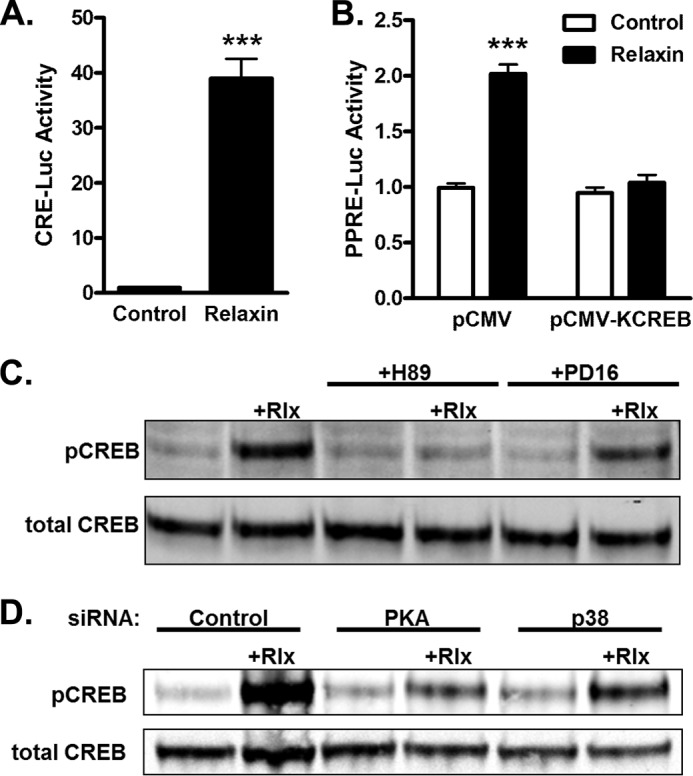
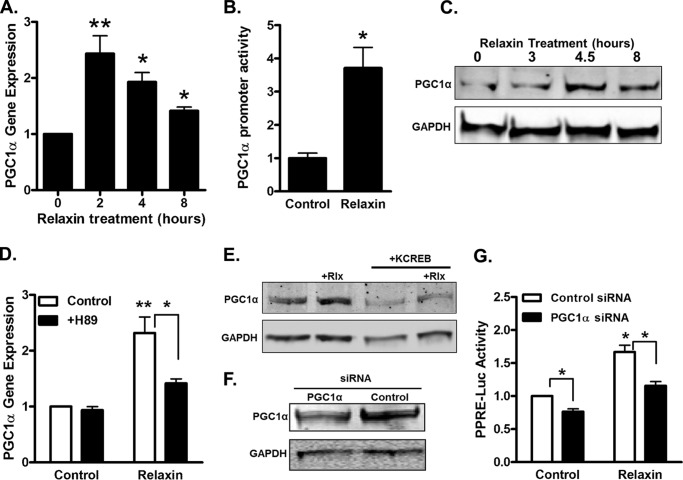
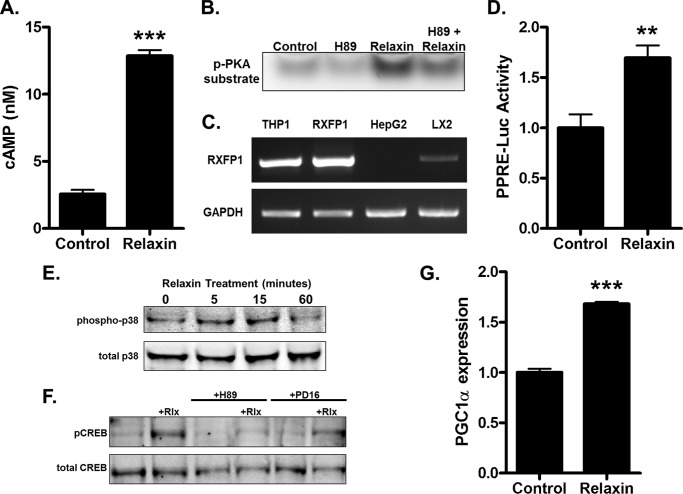
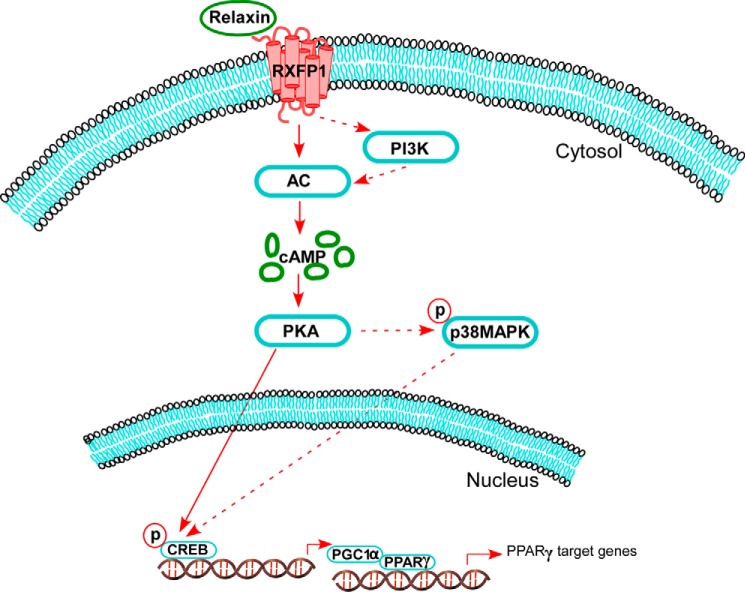
Relaxin activation of its receptor RXFP1 triggers multiple signaling pathways. Previously, we have shown that relaxin activates PPARγ transcriptional activity in a ligand-independent manner, but the mechanism for this effect was unknown. In this study, we examined the signaling pathways of downstream of RXFP1 leading to PPARγ activation. Using cells stably expressing RXFP1, we found that relaxin regulation of PPARγ activity requires accumulation of cAMP and subsequent activation of cAMP-dependent protein kinase (PKA). The activated PKA subsequently phosphorylated cAMP response element-binding protein (CREB) at Ser-133 to activate it directly, as well as indirectly through mitogen activated protein kinase p38 MAPK. Activated CREB was required for relaxin stimulation of PPARγ activity, while there was no evidence for a role of the nitric oxide or ERK MAPK pathways. Relaxin increased the mRNA and protein levels of the coactivator protein PGC1α, and this effect was dependent on PKA, and was completely abrogated by a dominant-negative form of CREB. This mechanism was confirmed in a hepatic stellate cell line stably that endogenously expresses RXFP1. Reduction of PGC1α levels using siRNA diminished the regulation of PPARγ by relaxin. These results suggest that relaxin activates the cAMP/PKA and p38 MAPK pathways to phosphorylate CREB, resulting in increased PGC1α levels. This provides a mechanism for the ligand-independent activation of PPARγ in response to relaxin.
Keywords: Peroxisome Proliferator-activated Receptor γ Coactivator 1-α (PGC-1a)(PPARGC1A); Peroxisome Proliferator-activated receptor (PPAR); Protein Kinase A (PKA); Relaxin; Relaxin Family Peptide Receptor 1 (RXFP1); cAMP Response Element-binding Protein (CREB); p38 MAPK.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
G Protein-coupled Receptor 40 (GPR40) and Peroxisome Proliferator-activated Receptor γ (PPARγ): AN INTEGRATED TWO-RECEPTOR SIGNALING PATHWAY.J Biol Chem. 2015 Aug 7;290(32):19544-57. doi: 10.1074/jbc.M115.638924. Epub 2015 Jun 23. J Biol Chem. 2015. PMID: 26105050 Free PMC article.
-
Relaxin signaling activates peroxisome proliferator-activated receptor gamma.Mol Cell Endocrinol. 2010 Feb 5;315(1-2):239-45. doi: 10.1016/j.mce.2009.08.014. Epub 2009 Aug 25. Mol Cell Endocrinol. 2010. PMID: 19712722 Free PMC article.
-
The cAMP signalling pathway activates CREB through PKA, p38 and MSK1 in NIH 3T3 cells.Cell Signal. 2005 Nov;17(11):1343-51. doi: 10.1016/j.cellsig.2005.02.003. Epub 2005 Mar 16. Cell Signal. 2005. PMID: 16125054
-
PPARγ signaling and emerging opportunities for improved therapeutics.Pharmacol Res. 2016 Sep;111:76-85. doi: 10.1016/j.phrs.2016.02.028. Epub 2016 Jun 4. Pharmacol Res. 2016. PMID: 27268145 Free PMC article. Review.
-
Natural products, PGC-1 α , and Duchenne muscular dystrophy.Acta Pharm Sin B. 2020 May;10(5):734-745. doi: 10.1016/j.apsb.2020.01.001. Epub 2020 Jan 8. Acta Pharm Sin B. 2020. PMID: 32528825 Free PMC article. Review.
Cited by
-
Anti-fibrotic actions of relaxin.Br J Pharmacol. 2017 May;174(10):962-976. doi: 10.1111/bph.13529. Epub 2016 Jul 7. Br J Pharmacol. 2017. PMID: 27250825 Free PMC article. Review.
-
Communication between mitochondria and other organelles: a brand-new perspective on mitochondria in cancer.Cell Biosci. 2019 Mar 19;9:27. doi: 10.1186/s13578-019-0289-8. eCollection 2019. Cell Biosci. 2019. PMID: 30931098 Free PMC article. Review.
-
Secretory products of the corpus luteum and preeclampsia.Hum Reprod Update. 2021 Jun 22;27(4):651-672. doi: 10.1093/humupd/dmab003. Hum Reprod Update. 2021. PMID: 33748839 Free PMC article. Review.
-
FOXO1 inhibition prevents renal ischemia-reperfusion injury via cAMP-response element binding protein/PPAR-γ coactivator-1α-mediated mitochondrial biogenesis.Br J Pharmacol. 2020 Jan;177(2):432-448. doi: 10.1111/bph.14878. Epub 2019 Dec 23. Br J Pharmacol. 2020. PMID: 31655022 Free PMC article.
-
Abscisic acid interplays with PPARγ receptors and ameliorates diabetes-induced cognitive deficits in rats.Avicenna J Phytomed. 2021 May-Jun;11(3):247-257. Avicenna J Phytomed. 2021. PMID: 34046321 Free PMC article.
References
-
- Sherwood O. D. (2004) Relaxin's Physiological Roles and Other Diverse Actions. Endocr. Rev. 25, 205–234 - PubMed
-
- Bathgate R. A., Halls M. L., van der Westhuizen E. T., Callander G. E., Kocan M., Summers R. J. (2013) Relaxin family peptides and their receptors. Physiol. Rev. 93, 405–480 - PubMed
-
- Hsu S. Y., Nakabayashi K., Nishi S., Kumagai J., Kudo M., Sherwood O. D., Hsueh A. J. W. (2002) Activation of orphan receptors by the hormone relaxin. Science 295, 671–674 - PubMed
-
- Bathgate R. A., Ivell R., Sanborn B. M., Sherwood O. D., Summers R. J. (2006) International Union of Pharmacology LVII: Recommendations for the Nomenclature of Receptors for Relaxin Family Peptides. Pharmacol. Rev. 58, 7–31 - PubMed
-
- Halls M. L., Bathgate R. A., Summers R. J. (2006) Relaxin family peptide receptors RXFP1 and RXFP2 modulate cAMP signaling by distinct mechanisms. Mol. Pharmacol. 70, 214–226 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
