

High-resolution structure and mechanism of an F/V-hybrid rotor ring in a Na⁺-coupled ATP synthase
- PMID: 25381992
- PMCID: PMC4228694
- DOI: 10.1038/ncomms6286
High-resolution structure and mechanism of an F/V-hybrid rotor ring in a Na⁺-coupled ATP synthase
Abstract
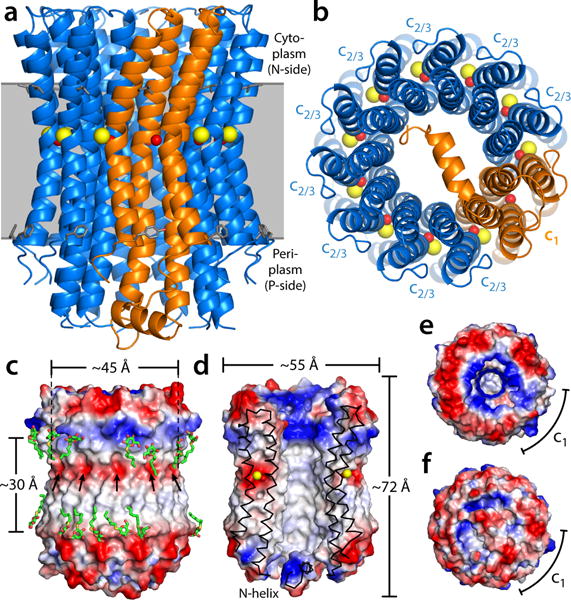
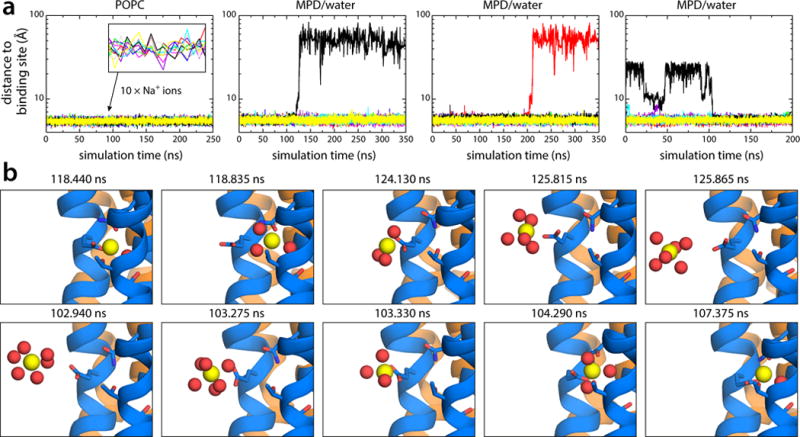
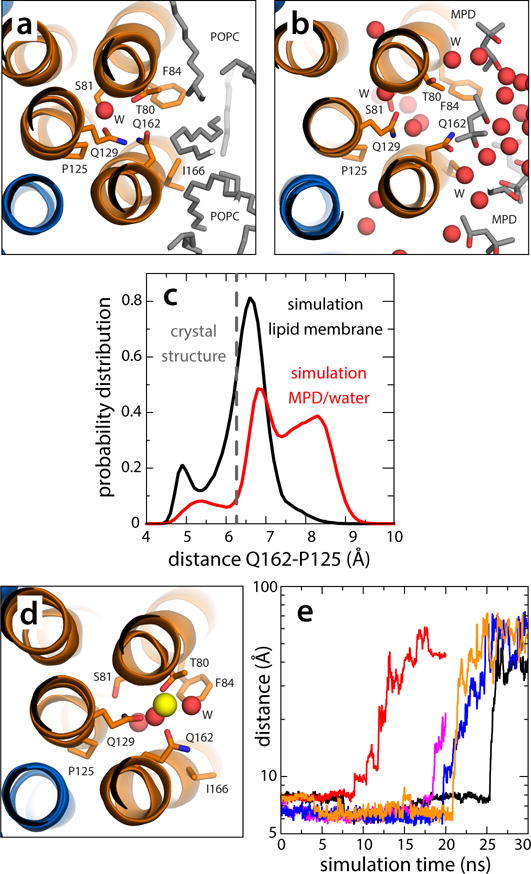
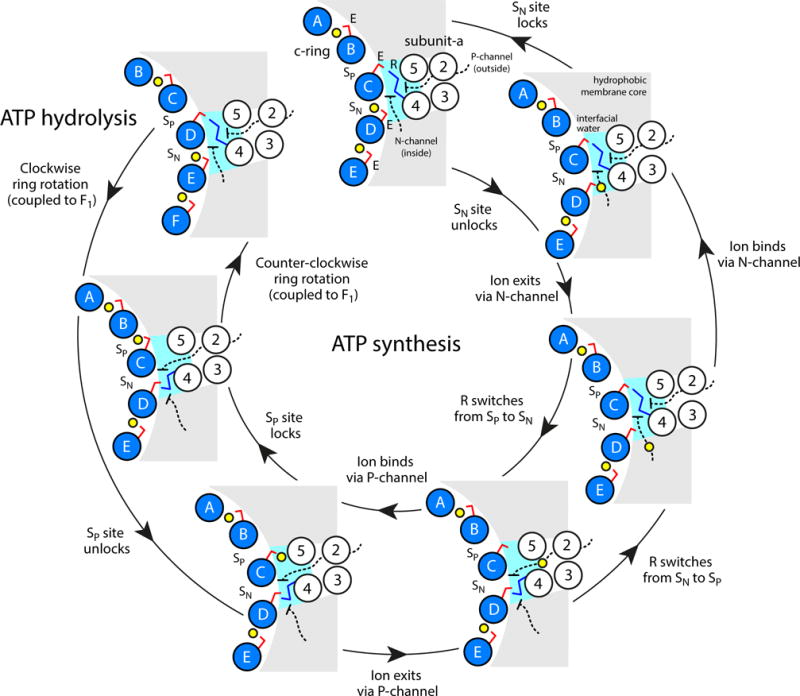
All rotary ATPases catalyse the interconversion of ATP and ADP-Pi through a mechanism that is coupled to the transmembrane flow of H(+) or Na(+). Physiologically, however, F/A-type enzymes specialize in ATP synthesis driven by downhill ion diffusion, while eukaryotic V-type ATPases function as ion pumps. To begin to rationalize the molecular basis for this functional differentiation, we solved the crystal structure of the Na(+)-driven membrane rotor of the Acetobacterium woodii ATP synthase, at 2.1 Å resolution. Unlike known structures, this rotor ring is a 9:1 heteromer of F- and V-type c-subunits and therefore features a hybrid configuration of ion-binding sites along its circumference. Molecular and kinetic simulations are used to dissect the mechanisms of Na(+) recognition and rotation of this c-ring, and to explain the functional implications of the V-type c-subunit. These structural and mechanistic insights indicate an evolutionary path between synthases and pumps involving adaptations in the rotor ring.
Conflict of interest statement
The authors declare no competing financial interests.
Figures




Similar articles
-
The structural features of Acetobacterium woodii F-ATP synthase reveal the importance of the unique subunit γ-loop in Na+ translocation and ATP synthesis.FEBS J. 2019 May;286(10):1894-1907. doi: 10.1111/febs.14793. Epub 2019 Mar 12. FEBS J. 2019. PMID: 30791207
-
An intermediate step in the evolution of ATPases: a hybrid F(0)-V(0) rotor in a bacterial Na(+) F(1)F(0) ATP synthase.FEBS J. 2008 May;275(9):1999-2007. doi: 10.1111/j.1742-4658.2008.06354.x. Epub 2008 Mar 19. FEBS J. 2008. PMID: 18355313
-
An intermediate step in the evolution of ATPases--the F1F0-ATPase from Acetobacterium woodii contains F-type and V-type rotor subunits and is capable of ATP synthesis.FEBS J. 2007 Jul;274(13):3421-8. doi: 10.1111/j.1742-4658.2007.05874.x. Epub 2007 Jun 6. FEBS J. 2007. PMID: 17555523
-
ATP synthases with novel rotor subunits: new insights into structure, function and evolution of ATPases.J Bioenerg Biomembr. 2005 Dec;37(6):455-60. doi: 10.1007/s10863-005-9491-y. J Bioenerg Biomembr. 2005. PMID: 16691483 Review.
-
Hybrid rotors in F1F(o) ATP synthases: subunit composition, distribution, and physiological significance.Biol Chem. 2015 Sep;396(9-10):1031-42. doi: 10.1515/hsz-2015-0137. Biol Chem. 2015. PMID: 25838297 Review.
Cited by
-
Lactate formation from fructose or C1 compounds in the acetogen Acetobacterium woodii by metabolic engineering.Appl Microbiol Biotechnol. 2023 Sep;107(17):5491-5502. doi: 10.1007/s00253-023-12637-7. Epub 2023 Jul 7. Appl Microbiol Biotechnol. 2023. PMID: 37417977 Free PMC article.
-
A new metabolic trait in an acetogen: Mixed acid fermentation of fructose in a methylene-tetrahydrofolate reductase mutant of Acetobacterium woodii.Environ Microbiol Rep. 2023 Oct;15(5):339-351. doi: 10.1111/1758-2229.13160. Epub 2023 May 7. Environ Microbiol Rep. 2023. PMID: 37150590 Free PMC article.
-
Reprogramming the metabolism of an acetogenic bacterium to homoformatogenesis.ISME J. 2023 Jul;17(7):984-992. doi: 10.1038/s41396-023-01411-2. Epub 2023 Apr 15. ISME J. 2023. PMID: 37061584 Free PMC article.
-
Direct observation of stepping rotation of V-ATPase reveals rigid component in coupling between Vo and V1 motors.Proc Natl Acad Sci U S A. 2022 Oct 18;119(42):e2210204119. doi: 10.1073/pnas.2210204119. Epub 2022 Oct 10. Proc Natl Acad Sci U S A. 2022. PMID: 36215468 Free PMC article.
-
Engineering Acetogenic Bacteria for Efficient One-Carbon Utilization.Front Microbiol. 2022 May 9;13:865168. doi: 10.3389/fmicb.2022.865168. eCollection 2022. Front Microbiol. 2022. PMID: 35615514 Free PMC article. Review.
References
-
- Junge W, Nelson N. Nature’s rotary electromotors. Science. 2005;308:642–644. - PubMed
-
- Fillingame RH. Coupling H+ transport and ATP synthesis in F1Fo-ATP synthases: glimpses of interacting parts in a dynamic molecular machine. J Exp Biol. 1997;200:217–224. - PubMed
-
- Hirata T, Nakamura N, Omote H, Wada Y, Futai M. Regulation and reversibility of vacuolar H+-ATPase. J Biol Chem. 2000;275:386–389. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
