

IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies
- PMID: 25371430
- PMCID: PMC4271533
- DOI: 10.1093/molbev/msu300
IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies
Abstract
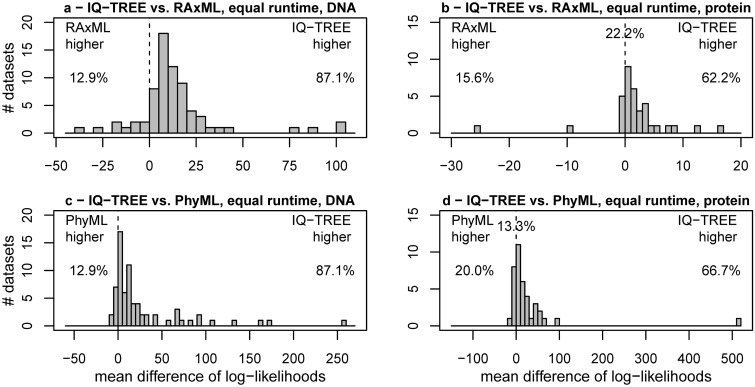
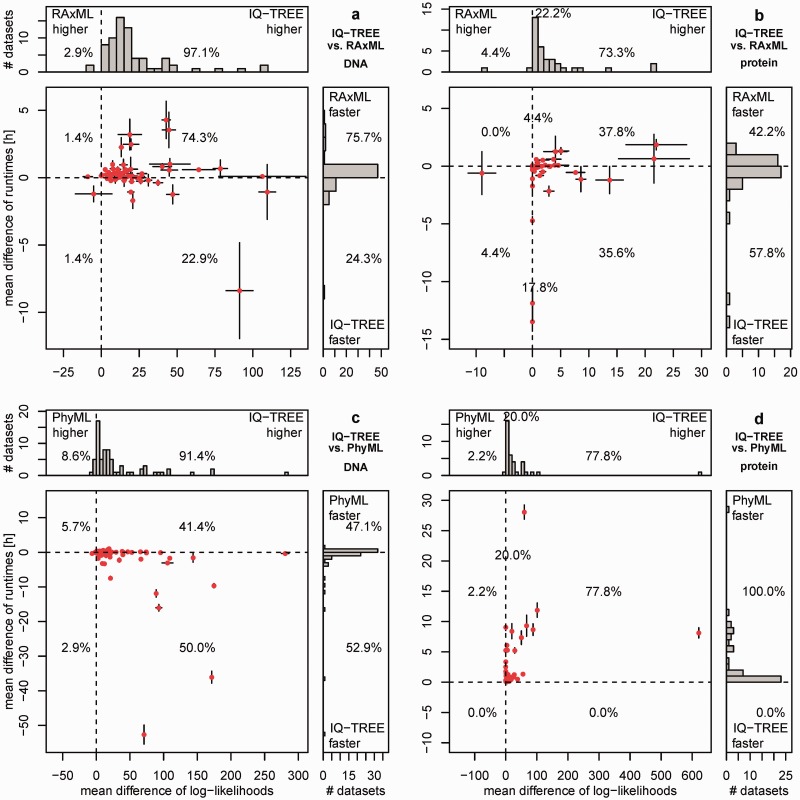
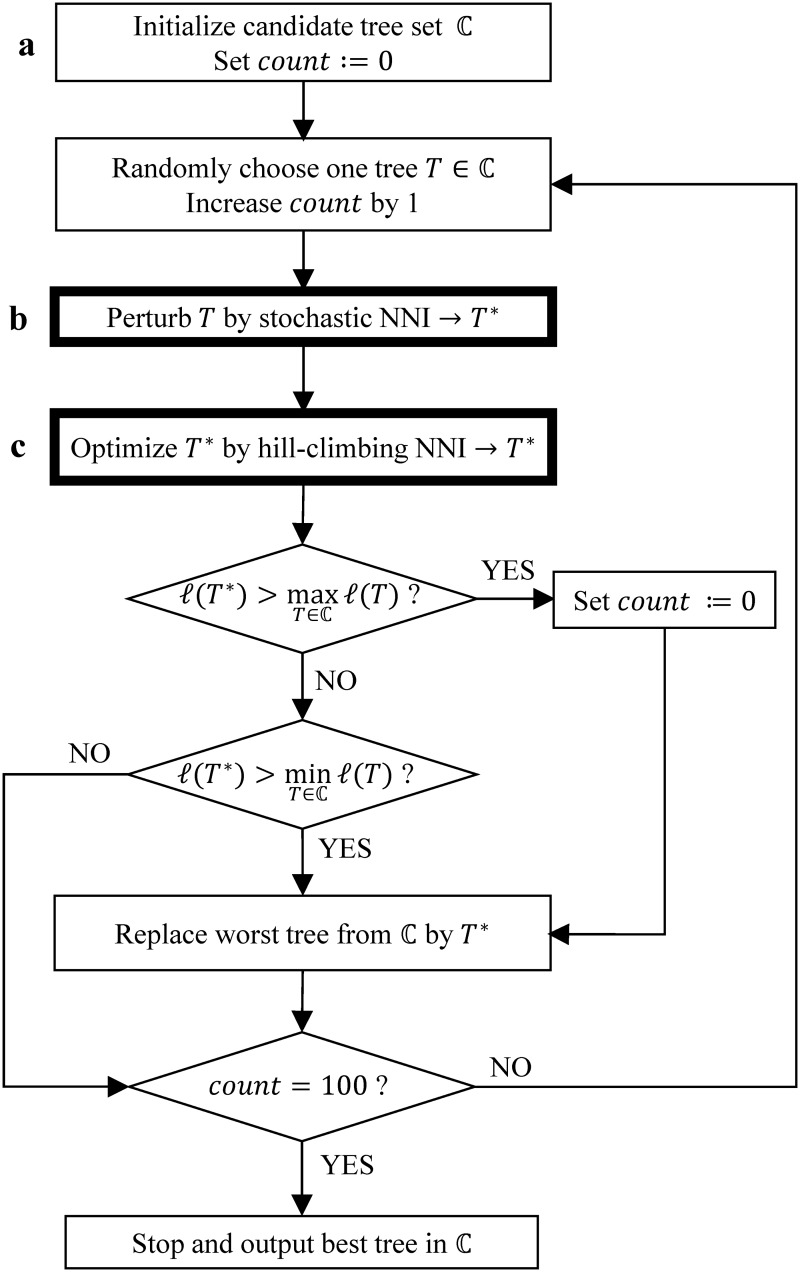
Large phylogenomics data sets require fast tree inference methods, especially for maximum-likelihood (ML) phylogenies. Fast programs exist, but due to inherent heuristics to find optimal trees, it is not clear whether the best tree is found. Thus, there is need for additional approaches that employ different search strategies to find ML trees and that are at the same time as fast as currently available ML programs. We show that a combination of hill-climbing approaches and a stochastic perturbation method can be time-efficiently implemented. If we allow the same CPU time as RAxML and PhyML, then our software IQ-TREE found higher likelihoods between 62.2% and 87.1% of the studied alignments, thus efficiently exploring the tree-space. If we use the IQ-TREE stopping rule, RAxML and PhyML are faster in 75.7% and 47.1% of the DNA alignments and 42.2% and 100% of the protein alignments, respectively. However, the range of obtaining higher likelihoods with IQ-TREE improves to 73.3-97.1%. IQ-TREE is freely available at http://www.cibiv.at/software/iqtree.
Keywords: maximum likelihood; phylogenetic inference; phylogeny; stochastic algorithm.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
Similar articles
-
Evaluating Fast Maximum Likelihood-Based Phylogenetic Programs Using Empirical Phylogenomic Data Sets.Mol Biol Evol. 2018 Feb 1;35(2):486-503. doi: 10.1093/molbev/msx302. Mol Biol Evol. 2018. PMID: 29177474 Free PMC article.
-
morePhyML: improving the phylogenetic tree space exploration with PhyML 3.Mol Phylogenet Evol. 2011 Dec;61(3):944-8. doi: 10.1016/j.ympev.2011.08.029. Epub 2011 Sep 8. Mol Phylogenet Evol. 2011. PMID: 21925283
-
RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference.Bioinformatics. 2019 Nov 1;35(21):4453-4455. doi: 10.1093/bioinformatics/btz305. Bioinformatics. 2019. PMID: 31070718 Free PMC article.
-
New approaches to phylogenetic tree search and their application to large numbers of protein alignments.Syst Biol. 2007 Oct;56(5):727-40. doi: 10.1080/10635150701611134. Syst Biol. 2007. PMID: 17849327
-
The Influence of the Number of Tree Searches on Maximum Likelihood Inference in Phylogenomics.Syst Biol. 2024 Oct 30;73(5):807-822. doi: 10.1093/sysbio/syae031. Syst Biol. 2024. PMID: 38940001
Cited by
-
Eustigmatophyte model of red-shifted chlorophyll a absorption in light-harvesting complexes.Commun Biol. 2024 Oct 29;7(1):1406. doi: 10.1038/s42003-024-07101-9. Commun Biol. 2024. PMID: 39472488 Free PMC article.
-
Characterization and phylogenetic analysis of the mitochondrial genome of Cylicostephanus longibursatus.Parasitol Res. 2024 Oct 29;123(10):363. doi: 10.1007/s00436-024-08385-w. Parasitol Res. 2024. PMID: 39467850
-
Plastid phylogenomics of Robinsonia (Senecioneae; Asteraceae), endemic to the Juan Fernández Islands: insights into structural organization and molecular evolution.BMC Plant Biol. 2024 Oct 28;24(1):1016. doi: 10.1186/s12870-024-05711-3. BMC Plant Biol. 2024. PMID: 39465373 Free PMC article.
-
The mitochondrial genome sequences of eleven leafhopper species of Batracomorphus (Hemiptera: Cicadellidae: Iassinae) reveal new gene rearrangements and phylogenetic implications.PeerJ. 2024 Oct 22;12:e18352. doi: 10.7717/peerj.18352. eCollection 2024. PeerJ. 2024. PMID: 39465150 Free PMC article.
-
Discovery of a new species of the subgenus Japonigekko (Squamata, Gekkonidae, Gekko) from the Hengduan Mountains, southwestern China: the best Japonigekko mountaineer.Zookeys. 2024 Oct 17;1215:289-309. doi: 10.3897/zookeys.1215.125043. eCollection 2024. Zookeys. 2024. PMID: 39464300 Free PMC article.
References
-
- Chor B, Tuller T. Maximum likelihood of evolutionary trees is hard. Lect Notes Comput Sci. 2005;3500:296–310.
-
- Farris JS. Methods for computing Wagner trees. Syst Zool. 1970;19:83–92.
-
- Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. - PubMed
-
- Felsenstein J. Inferring phylogenies. Sunderland (MA): Sinauer Associates; 2004.
-
- Fitch WM. Toward defining course of evolution—minimum change for a specific tree topology. Syst Zool. 1971;20:406–416.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
