

The nature of protein folding pathways
- PMID: 25326421
- PMCID: PMC4234557
- DOI: 10.1073/pnas.1411798111
The nature of protein folding pathways
Abstract
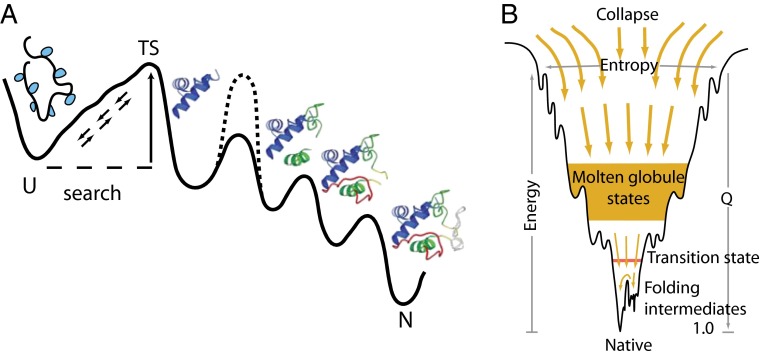
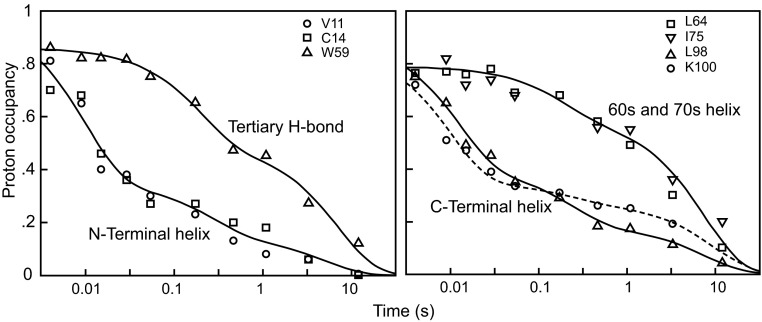
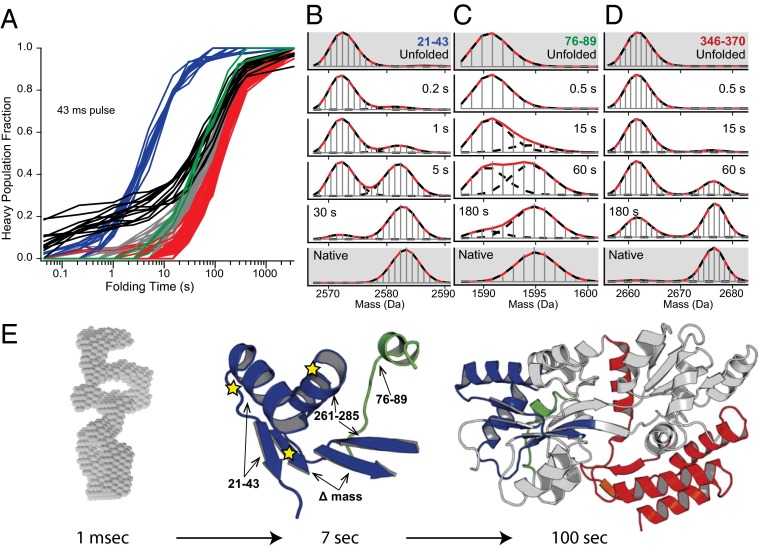
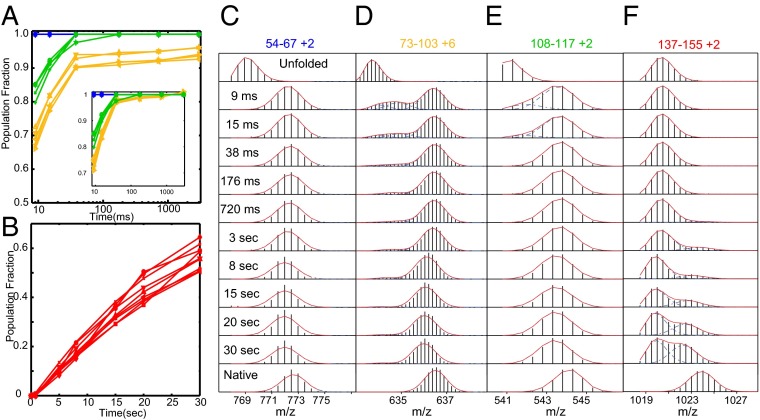
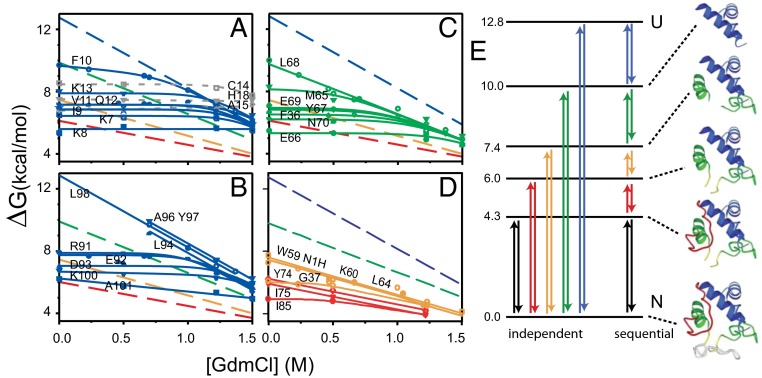
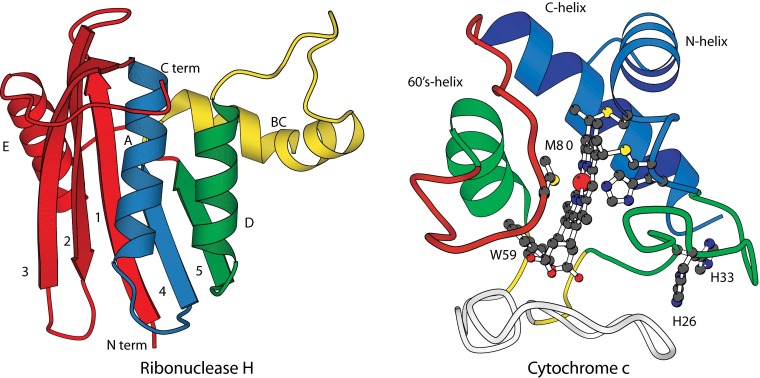
How do proteins fold, and why do they fold in that way? This Perspective integrates earlier and more recent advances over the 50-y history of the protein folding problem, emphasizing unambiguously clear structural information. Experimental results show that, contrary to prior belief, proteins are multistate rather than two-state objects. They are composed of separately cooperative foldon building blocks that can be seen to repeatedly unfold and refold as units even under native conditions. Similarly, foldons are lost as units when proteins are destabilized to produce partially unfolded equilibrium molten globules. In kinetic folding, the inherently cooperative nature of foldons predisposes the thermally driven amino acid-level search to form an initial foldon and subsequent foldons in later assisted searches. The small size of foldon units, ∼ 20 residues, resolves the Levinthal time-scale search problem. These microscopic-level search processes can be identified with the disordered multitrack search envisioned in the "new view" model for protein folding. Emergent macroscopic foldon-foldon interactions then collectively provide the structural guidance and free energy bias for the ordered addition of foldons in a stepwise pathway that sequentially builds the native protein. These conclusions reconcile the seemingly opposed new view and defined pathway models; the two models account for different stages of the protein folding process. Additionally, these observations answer the "how" and the "why" questions. The protein folding pathway depends on the same foldon units and foldon-foldon interactions that construct the native structure.
Keywords: hydrogen exchange; protein folding; protein structure.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Protein folding and misfolding: mechanism and principles.Q Rev Biophys. 2007 Nov;40(4):287-326. doi: 10.1017/S0033583508004654. Epub 2008 Apr 14. Q Rev Biophys. 2007. PMID: 18405419 Free PMC article. Review.
-
Protein folding: the stepwise assembly of foldon units.Proc Natl Acad Sci U S A. 2005 Mar 29;102(13):4741-6. doi: 10.1073/pnas.0501043102. Epub 2005 Mar 17. Proc Natl Acad Sci U S A. 2005. PMID: 15774579 Free PMC article.
-
The case for defined protein folding pathways.Proc Natl Acad Sci U S A. 2017 Aug 1;114(31):8253-8258. doi: 10.1073/pnas.1706196114. Epub 2017 Jun 19. Proc Natl Acad Sci U S A. 2017. PMID: 28630329 Free PMC article.
-
Cytochrome c folds through foldon-dependent native-like intermediates in an ordered pathway.Proc Natl Acad Sci U S A. 2016 Apr 5;113(14):3809-14. doi: 10.1073/pnas.1522674113. Epub 2016 Mar 10. Proc Natl Acad Sci U S A. 2016. PMID: 26966231 Free PMC article.
-
Protein Folding-How and Why: By Hydrogen Exchange, Fragment Separation, and Mass Spectrometry.Annu Rev Biophys. 2016 Jul 5;45:135-52. doi: 10.1146/annurev-biophys-062215-011121. Epub 2016 Apr 27. Annu Rev Biophys. 2016. PMID: 27145881 Free PMC article. Review.
Cited by
-
Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments.Nat Methods. 2019 Jul;16(7):595-602. doi: 10.1038/s41592-019-0459-y. Epub 2019 Jun 27. Nat Methods. 2019. PMID: 31249422 Free PMC article. Review.
-
Evolution to alternative levels of stable diversity leaves areas of niche space unexplored.PLoS Comput Biol. 2021 Jul 28;17(7):e1008650. doi: 10.1371/journal.pcbi.1008650. eCollection 2021 Jul. PLoS Comput Biol. 2021. PMID: 34319970 Free PMC article.
-
Using chirality to probe the conformational dynamics and assembly of intrinsically disordered amyloid proteins.Sci Rep. 2017 Oct 2;7(1):12433. doi: 10.1038/s41598-017-10525-5. Sci Rep. 2017. PMID: 28970487 Free PMC article.
-
Light-induced unfolding and refolding of supramolecular polymer nanofibres.Nat Commun. 2017 May 10;8:15254. doi: 10.1038/ncomms15254. Nat Commun. 2017. PMID: 28488694 Free PMC article.
-
A conserved folding nucleus sculpts the free energy landscape of bacterial and archaeal orthologs from a divergent TIM barrel family.Proc Natl Acad Sci U S A. 2021 Apr 27;118(17):e2019571118. doi: 10.1073/pnas.2019571118. Proc Natl Acad Sci U S A. 2021. PMID: 33875592 Free PMC article.
References
-
- Bai Y, Englander JJ, Mayne L, Milne JS, Englander SW. Thermodynamic parameters from hydrogen exchange measurements. Methods Enzymol. 1995;259:344–356. - PubMed
-
- Bai Y, Englander SW. Future directions in folding: The multi-state nature of protein structure. Proteins. 1996;24(2):145–151. - PubMed
-
- Levinthal C. Are there pathways for protein folding. J Chim Phys. 1968;65:44–45.
-
- Levinthal C. Mossbauer Spectroscopy in Biological Systems. Proceedings University of Illinois Bulletin. University of Illinois Press; Urbana, IL: 1969. How to fold graciously; pp. 22–24.
-
- Luheshi LM, Crowther DC, Dobson CM. Protein misfolding and disease: From the test tube to the organism. Curr Opin Chem Biol. 2008;12(1):25–31. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
