

Trinucleotide expansion in disease: why is there a length threshold?
- PMID: 25282113
- PMCID: PMC4252851
- DOI: 10.1016/j.gde.2014.07.003
Trinucleotide expansion in disease: why is there a length threshold?
Erratum in
- Curr Opin Genet Dev. 2015 Feb;30:80
Abstract
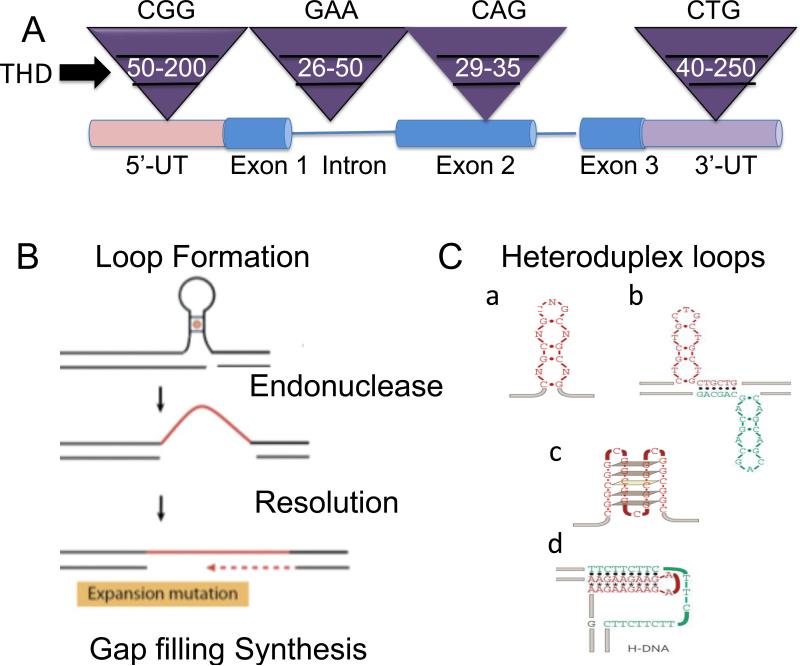
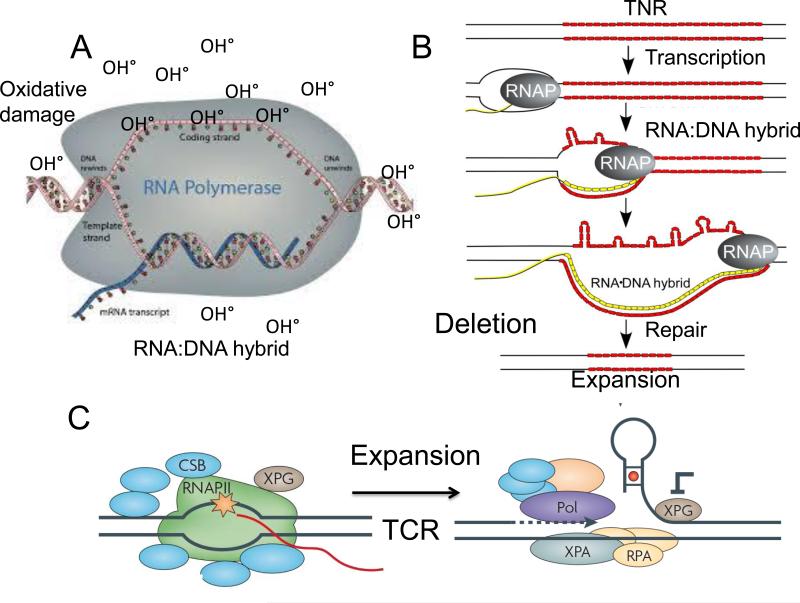
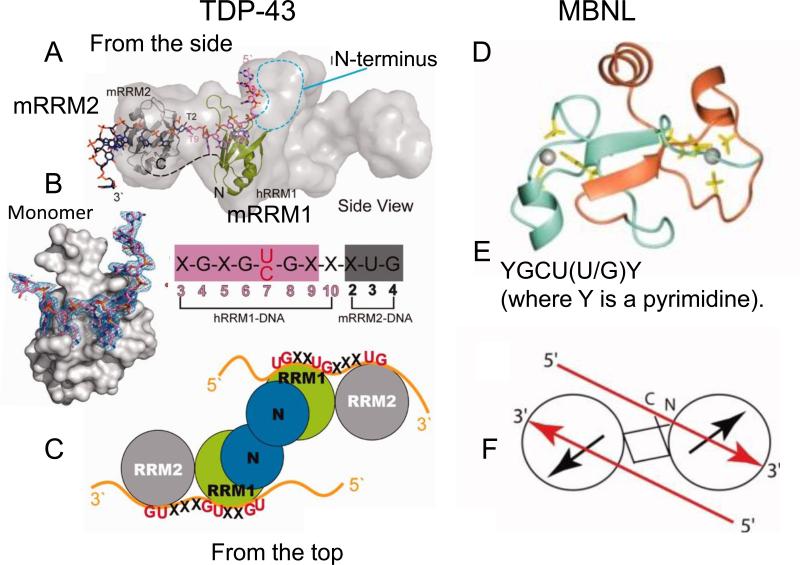
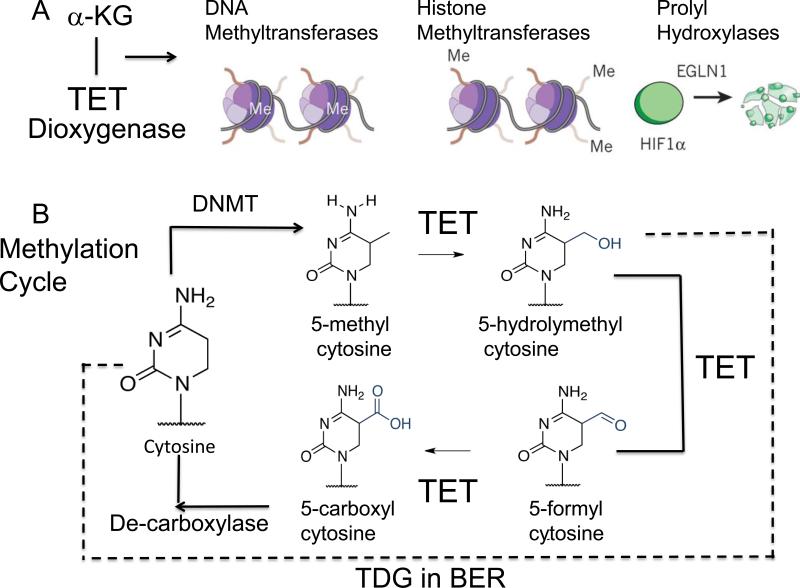
Trinucleotide repeats (TNRs) expansion disorders are severe neurodegenerative and neuromuscular disorders that arise from inheriting a long tract (30-50 copies) of a trinucleotide unit within or near an expressed gene (Figure 1a). The mutation is referred to as 'trinucleotide expansion' since the number of triplet units in a mutated gene is greater than the number found in the normal gene. Expansion becomes obvious once the number of repeating units passes a critical threshold length, but what happens at the threshold to render the repeating tract unstable? Here we discuss DNA-dependent and RNA-dependent models by which a particular DNA length permits a rapid transition to an unstable state.
Published by Elsevier Ltd.
Figures
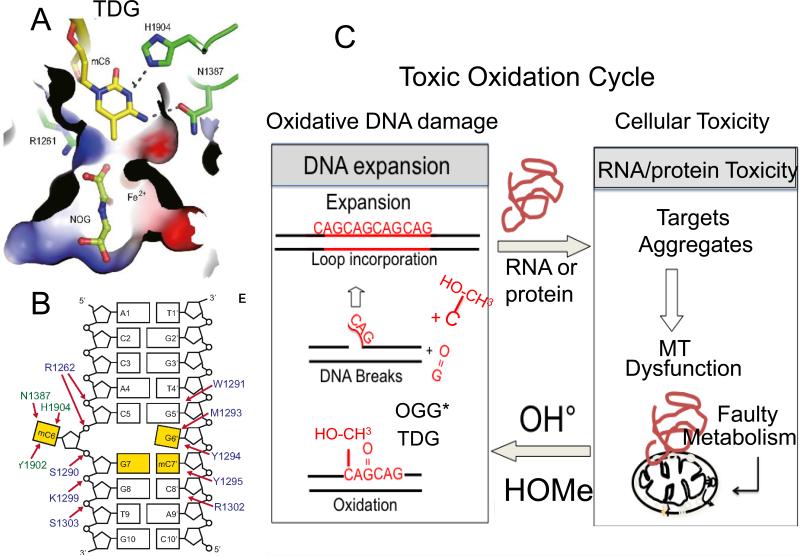
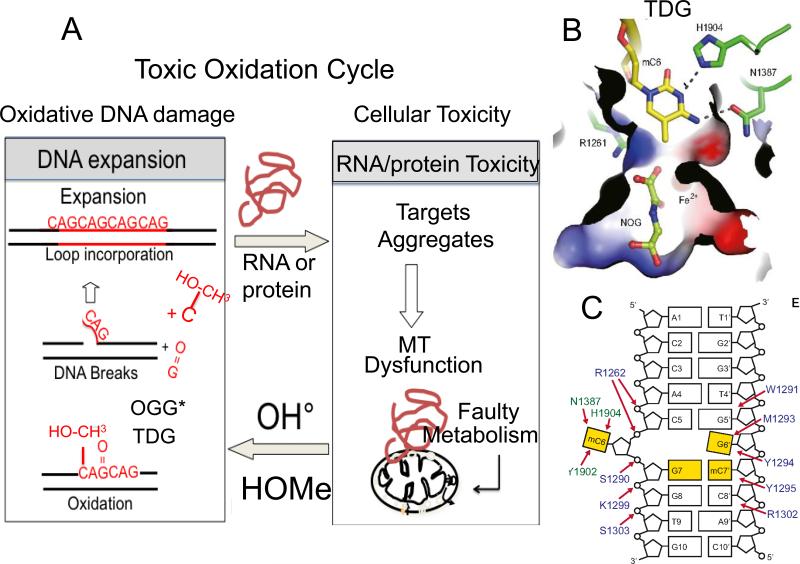
Similar articles
-
Progress on the mechanistic research of the trinucleotide repeat instabilities underlying human neurodegenerative diseases.Yi Chuan. 2021 Sep 20;43(9):835-848. doi: 10.16288/j.yczz.21-182. Yi Chuan. 2021. PMID: 34702697 Review.
-
Structural basis for triplet repeat disorders: a computational analysis.Bioinformatics. 1999 Nov;15(11):918-29. doi: 10.1093/bioinformatics/15.11.918. Bioinformatics. 1999. PMID: 10743558
-
Structural and Dynamical Properties of Nucleic Acid Hairpins Implicated in Trinucleotide Repeat Expansion Diseases.Biomolecules. 2024 Oct 10;14(10):1278. doi: 10.3390/biom14101278. Biomolecules. 2024. PMID: 39456210 Free PMC article. Review.
-
Transcription-induced DNA toxicity at trinucleotide repeats: double bubble is trouble.Cell Cycle. 2011 Feb 15;10(4):611-8. doi: 10.4161/cc.10.4.14729. Epub 2011 Feb 15. Cell Cycle. 2011. PMID: 21293182 Free PMC article.
-
Trinucleotide repeat expansion and neuropsychiatric disease.Arch Gen Psychiatry. 1999 Nov;56(11):1019-31. doi: 10.1001/archpsyc.56.11.1019. Arch Gen Psychiatry. 1999. PMID: 10565502 Review.
Cited by
-
Close encounters: Moving along bumps, breaks, and bubbles on expanded trinucleotide tracts.DNA Repair (Amst). 2017 Aug;56:144-155. doi: 10.1016/j.dnarep.2017.06.017. Epub 2017 Jun 9. DNA Repair (Amst). 2017. PMID: 28690053 Free PMC article. Review.
-
Tandem MutSβ binding to long extruded DNA trinucleotide repeats underpins pathogenic expansions.bioRxiv [Preprint]. 2023 Dec 13:2023.12.12.571350. doi: 10.1101/2023.12.12.571350. bioRxiv. 2023. PMID: 38168405 Free PMC article. Preprint.
-
Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions.Hum Genet. 2017 Sep;136(9):1247-1263. doi: 10.1007/s00439-017-1807-6. Epub 2017 May 8. Hum Genet. 2017. PMID: 28484853 Free PMC article. Review.
-
Stick-slip unfolding favors self-association of expanded HTT mRNA.bioRxiv [Preprint]. 2024 Jun 3:2024.05.31.596809. doi: 10.1101/2024.05.31.596809. bioRxiv. 2024. Update in: Nat Commun. 2024 Oct 9;15(1):8738. doi: 10.1038/s41467-024-52764-x. PMID: 38895475 Free PMC article. Updated. Preprint.
-
Polyglutamine Ataxias: Our Current Molecular Understanding and What the Future Holds for Antisense Therapies.Biomedicines. 2021 Oct 20;9(11):1499. doi: 10.3390/biomedicines9111499. Biomedicines. 2021. PMID: 34829728 Free PMC article. Review.
References
-
- McMurray CT. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet. 2010;11:786–99. [A comprehensive review of recent progress in linking the features of human disease with the replication and repair-dependent mechanisms of expansion and how they are used during different stages of development.] - PMC - PubMed
-
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–40. [An excellent review about the replication and repair-dependent mechanisms of triplet expansion.] - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
