

Genome analysis of the first Marseilleviridae representative from Australia indicates that most of its genes contribute to virus fitness
- PMID: 25275139
- PMCID: PMC4249118
- DOI: 10.1128/JVI.02414-14
Genome analysis of the first Marseilleviridae representative from Australia indicates that most of its genes contribute to virus fitness
Abstract
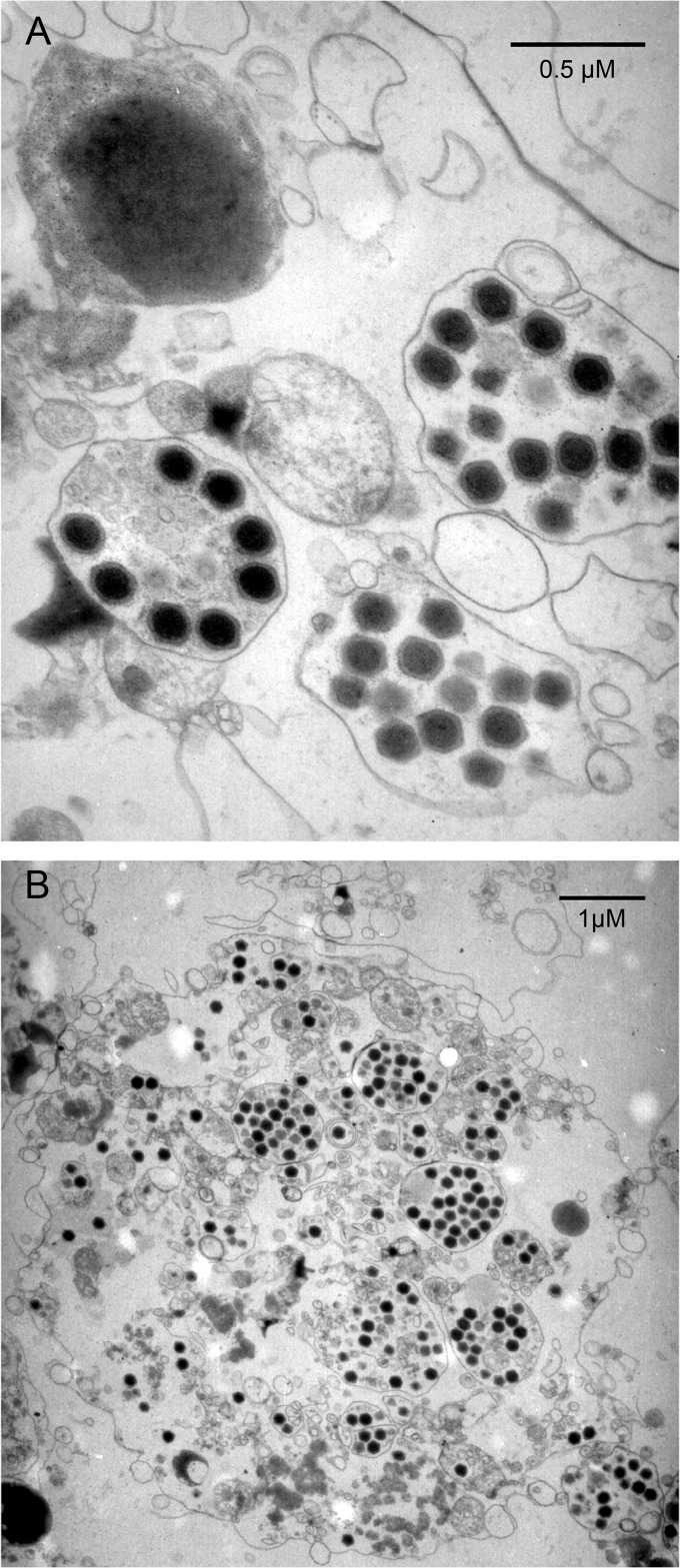
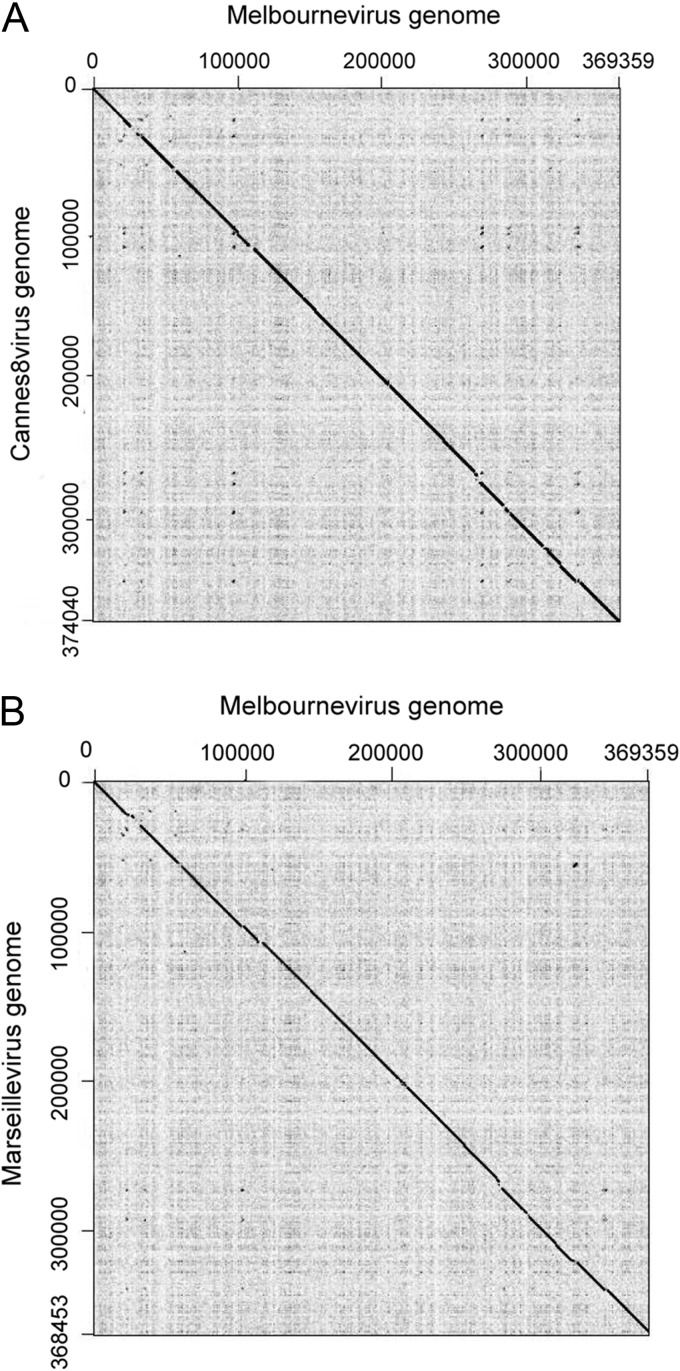
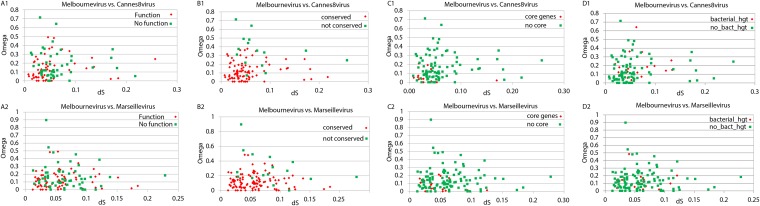
The family Marseilleviridae consists of Acanthamoeba-infecting large DNA viruses with icosahedral particles ∼ 0.2 μm in diameter and genome sizes in the 346- to 380-kb range. Since the isolation of Marseillevirus from a cooling tower in Paris (France) in 2009, the family Marseilleviridae has expanded rapidly, with representatives from Europe and Africa. Five members have been fully sequenced that are distributed among 3 emerging Marseilleviridae lineages. One comprises Marseillevirus and Cannes 8 virus, another one includes Insectomime virus and Tunisvirus, and the third one corresponds to the more distant Lausannevirus. We now report the genomic characterization of Melbournevirus, the first representative of the Marseilleviridae isolated from a freshwater pond in Melbourne, Australia. Despite the large distance separating this sampling point from France, Melbournevirus is remarkably similar to Cannes 8 virus and Marseillevirus, with most orthologous genes exhibiting more than 98% identical nucleotide sequences. We took advantage of this optimal evolutionary distance to evaluate the selection pressure, expressed as the ratio of nonsynonymous to synonymous mutations for various categories of genes. This ratio was found to be less than 1 for all of them, including those shared solely by the closest Melbournevirus and Cannes 8 virus isolates and absent from Lausannevirus. This suggests that most of the 403 protein-coding genes composing the large Melbournevirus genome are under negative/purifying selection and must thus significantly contribute to virus fitness. This conclusion contrasts with the more common view that many of the genes of the usually more diverse large DNA viruses might be (almost) dispensable.
Importance: A pervasive view is that viruses are fast-evolving parasites and carry the smallest possible amount of genomic information required to highjack the host cell machinery and perform their replication. This notion, probably inherited from the study of RNA viruses, is being gradually undermined by the discovery of DNA viruses with increasingly large gene content. These viruses also encode a variety of DNA repair functions, presumably slowing down their evolution by preserving their genomes from random alterations. On the other hand, these viruses also encode a majority of proteins without cellular homologs, including many shared only between the closest members of the same family. One may thus question the actual contribution of these anonymous and/or quasi-orphan genes to virus fitness. Genomic comparisons of Marseilleviridae, including a new Marseillevirus isolated in Australia, demonstrate that most of their genes, irrespective of their functions and conservation across families, are evolving under negative selection.
Copyright © 2014, American Society for Microbiology. All Rights Reserved.
Figures

Similar articles
-
Morphological and Taxonomic Properties of Tokyovirus, the First Marseilleviridae Member Isolated from Japan.Microbes Environ. 2016 Dec 23;31(4):442-448. doi: 10.1264/jsme2.ME16107. Epub 2016 Nov 19. Microbes Environ. 2016. PMID: 27867160 Free PMC article.
-
A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae.Viruses. 2016 Mar 10;8(3):76. doi: 10.3390/v8030076. Viruses. 2016. PMID: 26978387 Free PMC article.
-
Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes.Viruses. 2020 Nov 7;12(11):1270. doi: 10.3390/v12111270. Viruses. 2020. PMID: 33171839 Free PMC article.
-
The expanding family Marseilleviridae.Virology. 2014 Oct;466-467:27-37. doi: 10.1016/j.virol.2014.07.014. Epub 2014 Aug 5. Virology. 2014. PMID: 25104553 Review.
-
Evolutionary genomics of nucleo-cytoplasmic large DNA viruses.Virus Res. 2006 Apr;117(1):156-84. doi: 10.1016/j.virusres.2006.01.009. Epub 2006 Feb 21. Virus Res. 2006. PMID: 16494962 Review.
Cited by
-
The Astounding World of Glycans from Giant Viruses.Chem Rev. 2022 Oct 26;122(20):15717-15766. doi: 10.1021/acs.chemrev.2c00118. Epub 2022 Jul 12. Chem Rev. 2022. PMID: 35820164 Free PMC article. Review.
-
Comparative Genomics of Chrysochromulina Ericina Virus and Other Microalga-Infecting Large DNA Viruses Highlights Their Intricate Evolutionary Relationship with the Established Mimiviridae Family.J Virol. 2017 Jun 26;91(14):e00230-17. doi: 10.1128/JVI.00230-17. Print 2017 Jul 15. J Virol. 2017. PMID: 28446675 Free PMC article.
-
In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba.Proc Natl Acad Sci U S A. 2015 Sep 22;112(38):E5327-35. doi: 10.1073/pnas.1510795112. Epub 2015 Sep 8. Proc Natl Acad Sci U S A. 2015. PMID: 26351664 Free PMC article.
-
Amoebae, Giant Viruses, and Virophages Make Up a Complex, Multilayered Threesome.Front Cell Infect Microbiol. 2018 Jan 11;7:527. doi: 10.3389/fcimb.2017.00527. eCollection 2017. Front Cell Infect Microbiol. 2018. PMID: 29376032 Free PMC article. Review.
-
The number of genes encoding repeat domain-containing proteins positively correlates with genome size in amoebal giant viruses.Virus Evol. 2018 Jan 3;4(1):vex039. doi: 10.1093/ve/vex039. eCollection 2018 Jan. Virus Evol. 2018. PMID: 29308275 Free PMC article.
References
-
- Legendre M, Audic S, Poirot O, Hingamp P, Seltzer V, Byrne D, Lartigue A, Lescot M, Bernadac A, Poulain J, Abergel C, Claverie JM. 2010. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 20:664–674. 10.1101/gr.102582.109. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
