

Functional annotation of colon cancer risk SNPs
- PMID: 25268989
- PMCID: PMC4200523
- DOI: 10.1038/ncomms6114
Functional annotation of colon cancer risk SNPs
Abstract
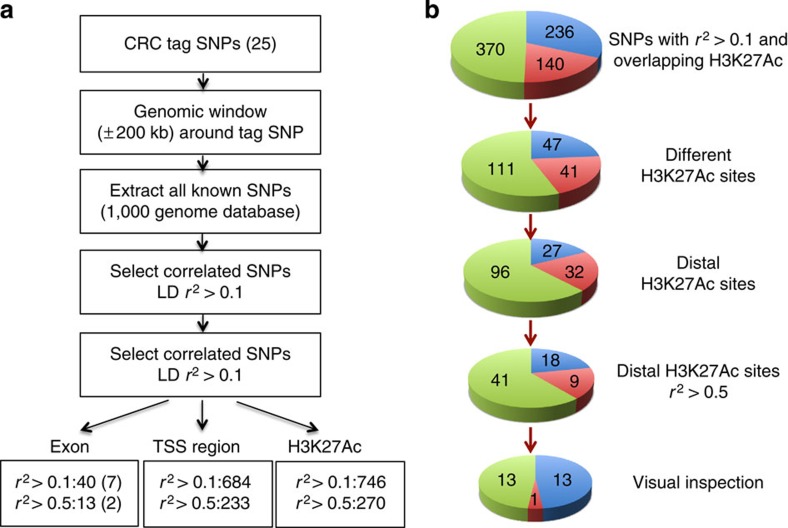
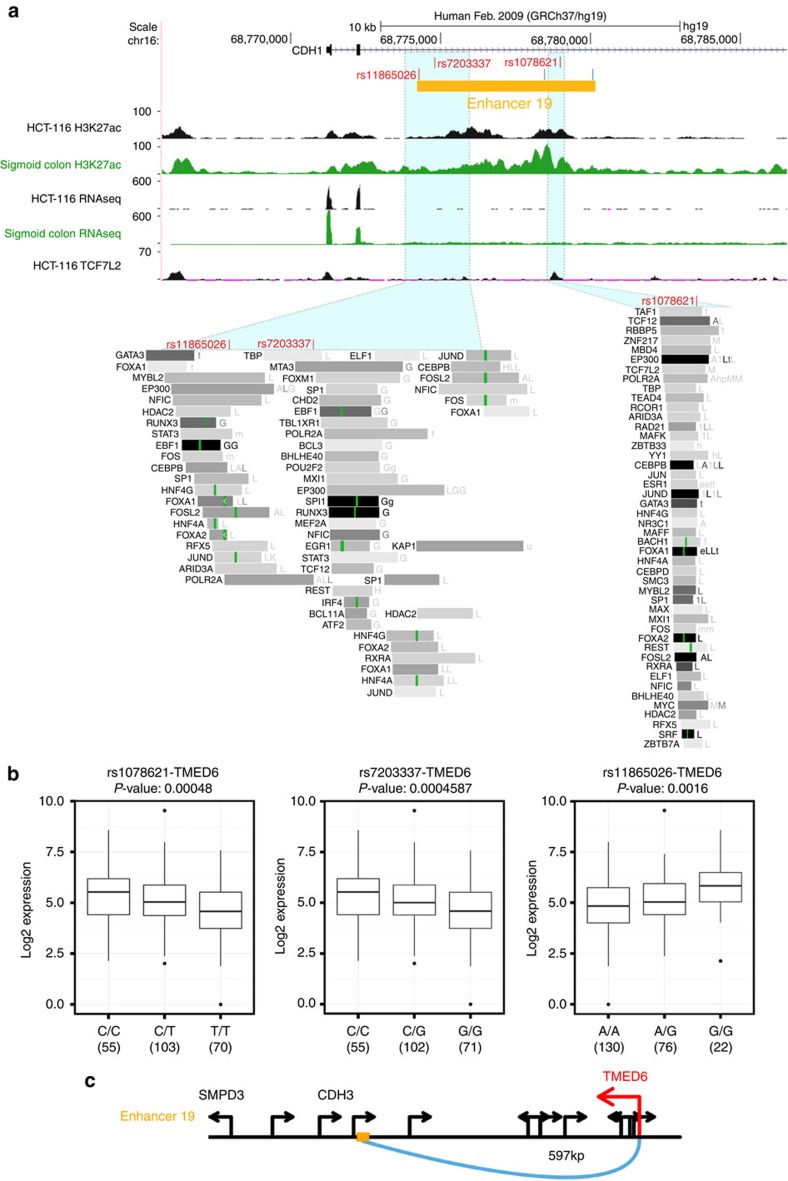
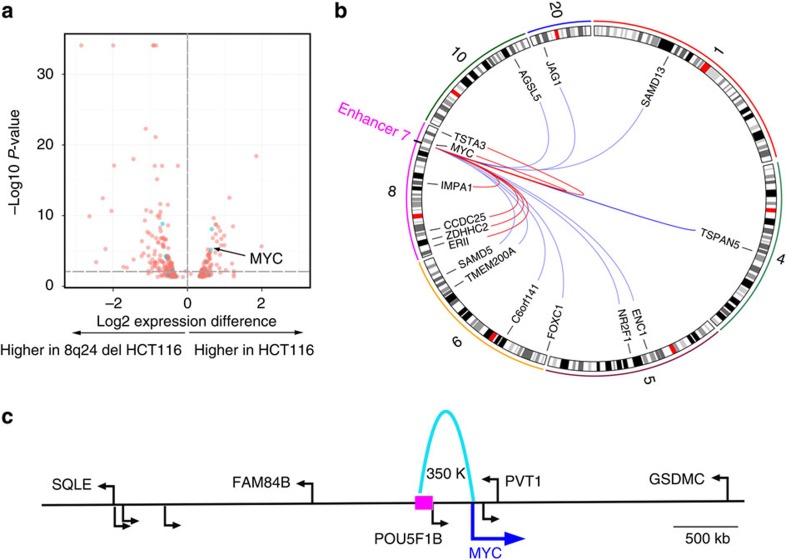
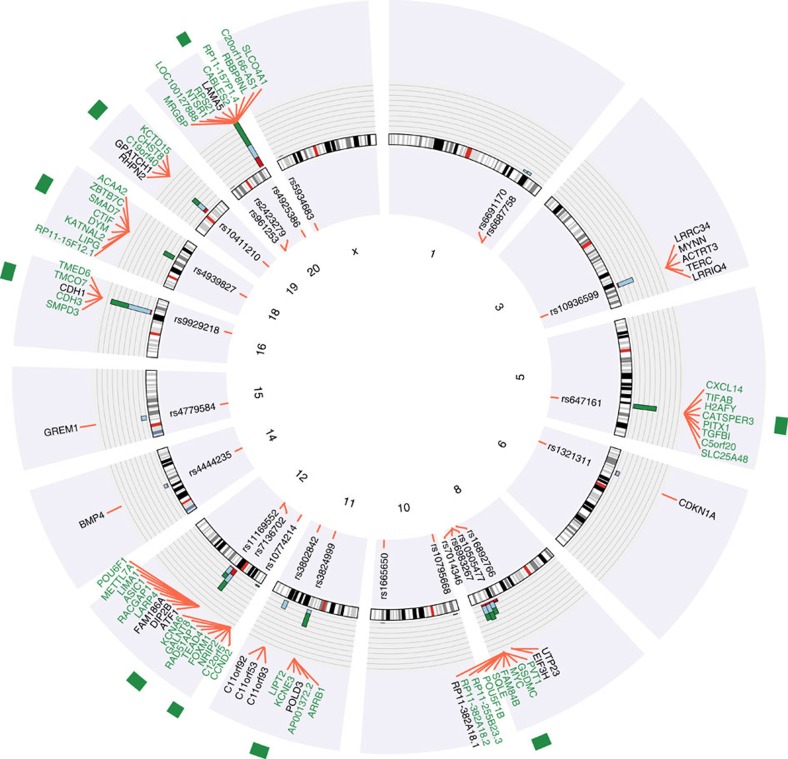
Colorectal cancer (CRC) is a leading cause of cancer-related deaths in the United States. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) associated with increased risk for CRC. A molecular understanding of the functional consequences of this genetic variation has been complicated because each GWAS SNP is a surrogate for hundreds of other SNPs, most of which are located in non-coding regions. Here we use genomic and epigenomic information to test the hypothesis that the GWAS SNPs and/or correlated SNPs are in elements that regulate gene expression, and identify 23 promoters and 28 enhancers. Using gene expression data from normal and tumour cells, we identify 66 putative target genes of the risk-associated enhancers (10 of which were also identified by promoter SNPs). Employing CRISPR nucleases, we delete one risk-associated enhancer and identify genes showing altered expression. We suggest that similar studies be performed to characterize all CRC risk-associated enhancers.
Figures

Similar articles
-
SNPs associated with colorectal cancer at 15q13.3 affect risk enhancers that modulate GREM1 gene expression.Hum Mutat. 2021 Mar;42(3):237-245. doi: 10.1002/humu.24166. Epub 2021 Feb 2. Hum Mutat. 2021. PMID: 33476087 Free PMC article.
-
Integrative analysis of liver-specific non-coding regulatory SNPs associated with the risk of coronary artery disease.Am J Hum Genet. 2021 Mar 4;108(3):411-430. doi: 10.1016/j.ajhg.2021.02.006. Epub 2021 Feb 23. Am J Hum Genet. 2021. PMID: 33626337 Free PMC article.
-
Genetic Predisposition to Colon and Rectal Adenocarcinoma Is Mediated by a Super-enhancer Polymorphism Coactivating CD9 and PLEKHG6.Cancer Epidemiol Biomarkers Prev. 2020 Apr;29(4):850-859. doi: 10.1158/1055-9965.EPI-19-1116. Epub 2020 Jan 27. Cancer Epidemiol Biomarkers Prev. 2020. PMID: 31988071
-
The identification of colon cancer susceptibility genes by using genome-wide scans.Methods Mol Biol. 2010;653:3-21. doi: 10.1007/978-1-60761-759-4_1. Methods Mol Biol. 2010. PMID: 20721734 Review.
-
Progress on functional mechanisms of colorectal cancer causal SNPs in post-GWAS.Yi Chuan. 2021 Mar 16;43(3):203-214. doi: 10.16288/j.yczz.20-320. Yi Chuan. 2021. PMID: 33724205 Review.
Cited by
-
The importance of detailed epigenomic profiling of different cell types within organs.Epigenomics. 2016 Jun;8(6):817-29. doi: 10.2217/epi-2016-0005. Epub 2016 Jun 15. Epigenomics. 2016. PMID: 27305639 Free PMC article. Review.
-
A CRISPR Path to Engineering New Genetic Mouse Models for Cardiovascular Research.Arterioscler Thromb Vasc Biol. 2016 Jun;36(6):1058-75. doi: 10.1161/ATVBAHA.116.304790. Epub 2016 Apr 21. Arterioscler Thromb Vasc Biol. 2016. PMID: 27102963 Free PMC article. Review.
-
Integrative Functional Annotation of 52 Genetic Loci Influencing Myocardial Mass Identifies Candidate Regulatory Variants and Target Genes.Circ Genom Precis Med. 2019 Feb;12(2):e002328. doi: 10.1161/CIRCGEN.118.002328. Circ Genom Precis Med. 2019. PMID: 30681347 Free PMC article.
-
Integrative omics for health and disease.Nat Rev Genet. 2018 May;19(5):299-310. doi: 10.1038/nrg.2018.4. Epub 2018 Feb 26. Nat Rev Genet. 2018. PMID: 29479082 Free PMC article. Review.
-
Comprehensive functional annotation of susceptibility SNPs prioritized 10 genes for schizophrenia.Transl Psychiatry. 2019 Jan 31;9(1):56. doi: 10.1038/s41398-019-0398-5. Transl Psychiatry. 2019. PMID: 30705251 Free PMC article.
References
-
- Fearon E. R. Molecular genetics of colorectal cancer. Annu. Rev. Pathol. 6, 479–507 (2011). - PubMed
-
- Manolio T. A. Genomewide association studies and assessment of the risk of disease. New Engl. J. Med. 363, 166–176 (2010). - PubMed
-
- Zanke B. W. et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet. 39, 989–994 (2007). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
