

STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity
- PMID: 25258038
- PMCID: PMC4181732
- DOI: 10.1186/s13023-014-0146-0
STUB1 mutations in autosomal recessive ataxias - evidence for mutation-specific clinical heterogeneity
Abstract
Background: A subset of hereditary cerebellar ataxias is inherited as autosomal recessive traits (ARCAs). Classification of recessive ataxias due to phenotypic differences in the cerebellum and cerebellar structures is constantly evolving due to new identified disease genes. Recently, reports have linked mutations in genes involved in ubiquitination (RNF216, OTUD4, STUB1) to ARCA with hypogonadism.
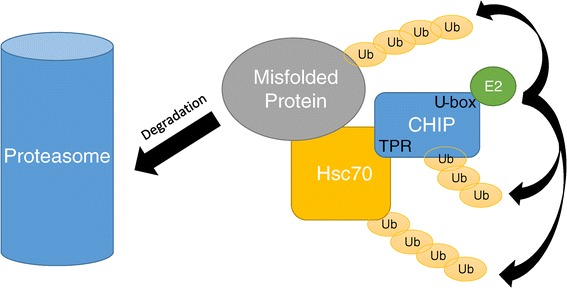
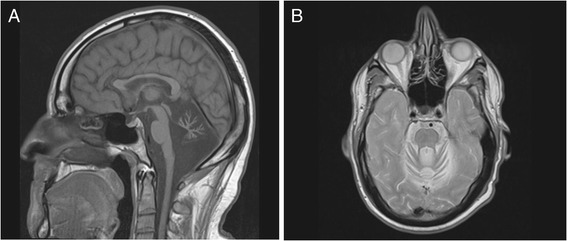
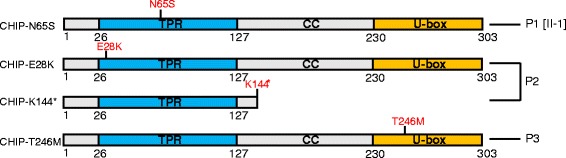
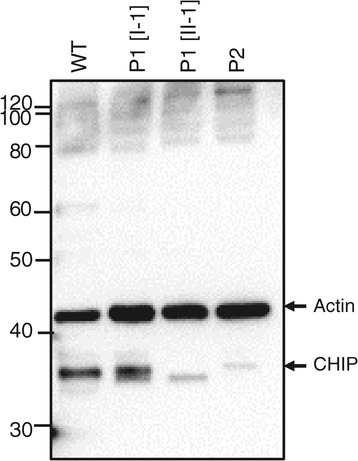
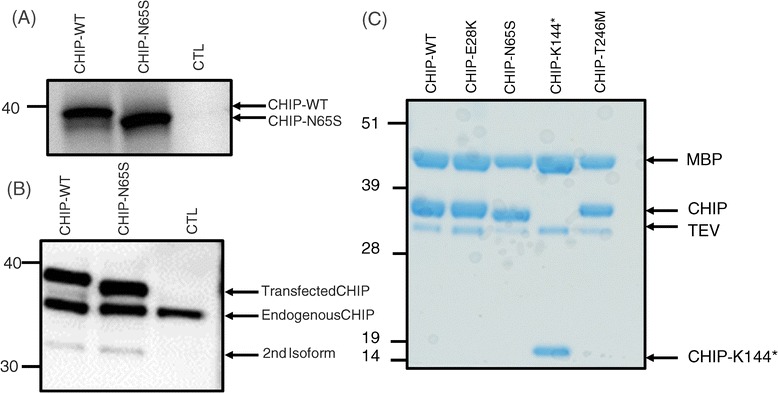
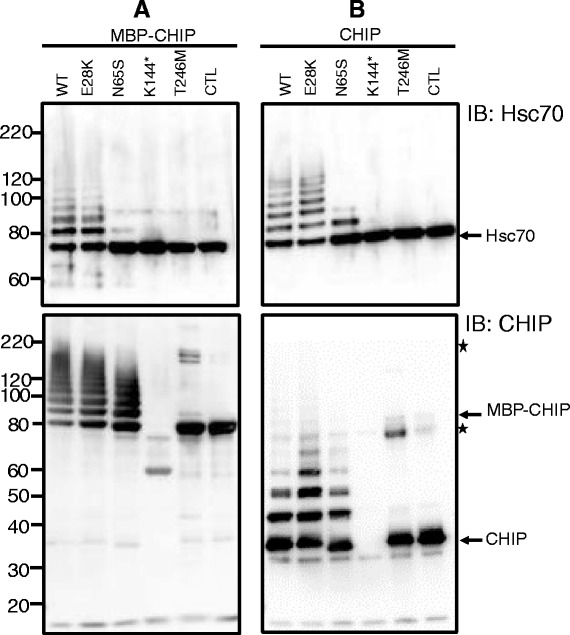
Methods and results: With a combination of homozygozity mapping and exome sequencing, we identified three mutations in STUB1 in two families with ARCA and cognitive impairment; a homozygous missense variant (c.194A > G, p.Asn65Ser) that segregated in three affected siblings, and a missense change (c.82G > A, p.Glu28Lys) which was inherited in trans with a nonsense mutation (c.430A > T, p.Lys144Ter) in another patient. STUB1 encodes CHIP (C-terminus of Heat shock protein 70 - Interacting Protein), a dual function protein with a role in ubiquitination as a co-chaperone with heat shock proteins, and as an E3 ligase. We show that the p.Asn65Ser substitution impairs CHIP's ability to ubiquitinate HSC70 in vitro, despite being able to self-ubiquitinate. These results are consistent with previous studies highlighting this as a critical residue for the interaction between CHIP and its co-chaperones. Furthermore, we show that the levels of CHIP are strongly reduced in vivo in patients' fibroblasts compared to controls.
Conclusions: These results suggest that STUB1 mutations might cause disease by impacting not only the E3 ligase function, but also its protein interaction properties and protein amount. Whether the clinical heterogeneity seen in STUB1 ARCA can be related to the location of the mutations remains to be understood, but interestingly, all siblings with the p.Asn65Ser substitution showed a marked appearance of accelerated aging not previously described in STUB1 related ARCA, none display hormonal aberrations/clinical hypogonadism while some affected family members had diabetes, alopecia, uveitis and ulcerative colitis, further refining the spectrum of STUB1 related disease.
Figures
Similar articles
-
Clinical and Genetic Characterization of Autosomal Recessive Spinocerebellar Ataxia Type 16 (SCAR16) in Taiwan.Cerebellum. 2020 Aug;19(4):544-549. doi: 10.1007/s12311-020-01136-4. Cerebellum. 2020. PMID: 32367277
-
Phenotype and frequency of STUB1 mutations: next-generation screenings in Caucasian ataxia and spastic paraplegia cohorts.Orphanet J Rare Dis. 2014 Apr 17;9:57. doi: 10.1186/1750-1172-9-57. Orphanet J Rare Dis. 2014. PMID: 24742043 Free PMC article.
-
Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP.Hum Mol Genet. 2014 Feb 15;23(4):1013-24. doi: 10.1093/hmg/ddt497. Epub 2013 Oct 9. Hum Mol Genet. 2014. PMID: 24113144 Free PMC article.
-
Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias.Hum Genome Var. 2014 Oct 23;1:14018. doi: 10.1038/hgv.2014.18. eCollection 2014. Hum Genome Var. 2014. PMID: 27081508 Free PMC article. Review.
-
Spinocerebellar ataxia type 48: last but not least.Neurol Sci. 2020 Sep;41(9):2423-2432. doi: 10.1007/s10072-020-04408-3. Epub 2020 Apr 27. Neurol Sci. 2020. PMID: 32342324 Review.
Cited by
-
Negative Regulation of the Innate Immune Response through Proteasomal Degradation and Deubiquitination.Viruses. 2021 Mar 30;13(4):584. doi: 10.3390/v13040584. Viruses. 2021. PMID: 33808506 Free PMC article. Review.
-
HSPB8 frameshift mutant aggregates weaken chaperone-assisted selective autophagy in neuromyopathies.Autophagy. 2023 Aug;19(8):2217-2239. doi: 10.1080/15548627.2023.2179780. Epub 2023 Feb 28. Autophagy. 2023. PMID: 36854646 Free PMC article.
-
Movement Disorders Associated with Hypogonadism.Mov Disord Clin Pract. 2021 Jul 29;8(7):997-1011. doi: 10.1002/mdc3.13308. eCollection 2021 Oct. Mov Disord Clin Pract. 2021. PMID: 34631935 Free PMC article. Review.
-
In vitro characterization of six STUB1 variants in spinocerebellar ataxia 16 reveals altered structural properties for the encoded CHIP proteins.Biosci Rep. 2017 Apr 28;37(2):BSR20170251. doi: 10.1042/BSR20170251. Print 2017 Apr 30. Biosci Rep. 2017. PMID: 28396517 Free PMC article.
-
Correspondence on "Clinical, neuropathological, and genetic characterization of STUB1 variants in cerebellar ataxias: a frequent cause of predominant cognitive impairment" by Roux et al.Genet Med. 2021 Jun;23(6):1171-1172. doi: 10.1038/s41436-021-01104-1. Epub 2021 Feb 9. Genet Med. 2021. PMID: 33564152 Free PMC article. No abstract available.
References
-
- Shi CH, Schisler JC, Rubel CE, Tan S, Song B, McDonough H, Xu L, Portbury AL, Mao CY, True C, Wang RH, Wang QZ, Sun SL, Seminara SB, Patterson C, Xu YM. Ataxia and hypogonadism caused by the loss of ubiquitin ligase activity of the U box protein CHIP. Hum Mol Genet. 2014;23:1013–1024. doi: 10.1093/hmg/ddt497. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
