

Conformation-independent structural comparison of macromolecules with ProSMART
- PMID: 25195761
- PMCID: PMC4157452
- DOI: 10.1107/S1399004714016241
Conformation-independent structural comparison of macromolecules with ProSMART
Abstract
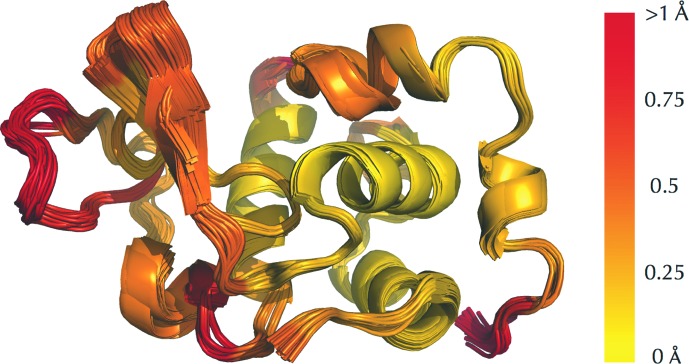
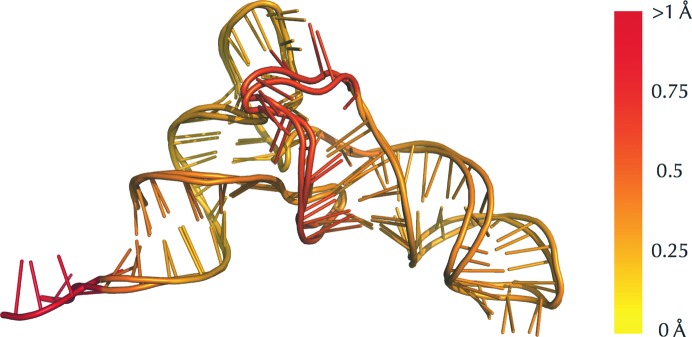
The identification and exploration of (dis)similarities between macromolecular structures can help to gain biological insight, for instance when visualizing or quantifying the response of a protein to ligand binding. Obtaining a residue alignment between compared structures is often a prerequisite for such comparative analysis. If the conformational change of the protein is dramatic, conventional alignment methods may struggle to provide an intuitive solution for straightforward analysis. To make such analyses more accessible, the Procrustes Structural Matching Alignment and Restraints Tool (ProSMART) has been developed, which achieves a conformation-independent structural alignment, as well as providing such additional functionalities as the generation of restraints for use in the refinement of macromolecular models. Sensible comparison of protein (or DNA/RNA) structures in the presence of conformational changes is achieved by enforcing neither chain nor domain rigidity. The visualization of results is facilitated by popular molecular-graphics software such as CCP4mg and PyMOL, providing intuitive feedback regarding structural conservation and subtle dissimilarities between close homologues that can otherwise be hard to identify. Automatically generated colour schemes corresponding to various residue-based scores are provided, which allow the assessment of the conservation of backbone and side-chain conformations relative to the local coordinate frame. Structural comparison tools such as ProSMART can help to break the complexity that accompanies the constantly growing pool of structural data into a more readily accessible form, potentially offering biological insight or influencing subsequent experiments.
Keywords: ProSMART; Procrustes; alignment; external restraints; refinement; structural comparison.
Figures
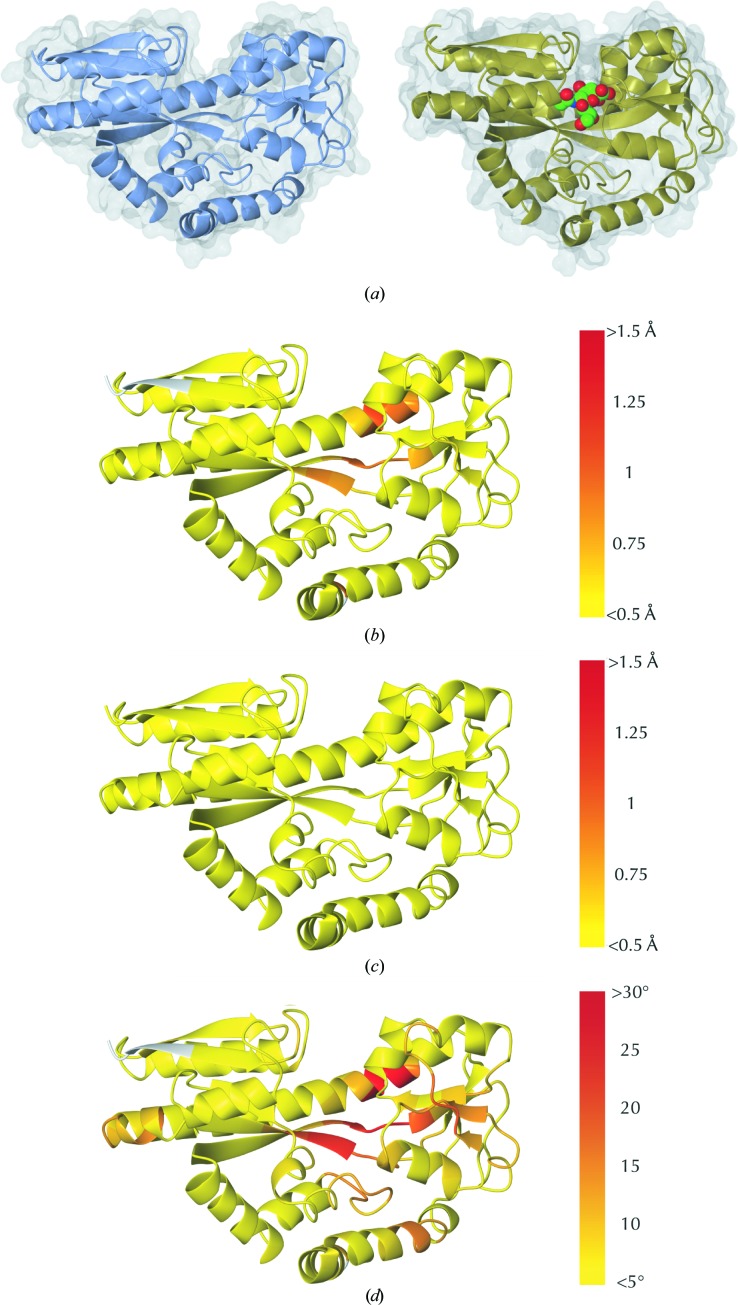
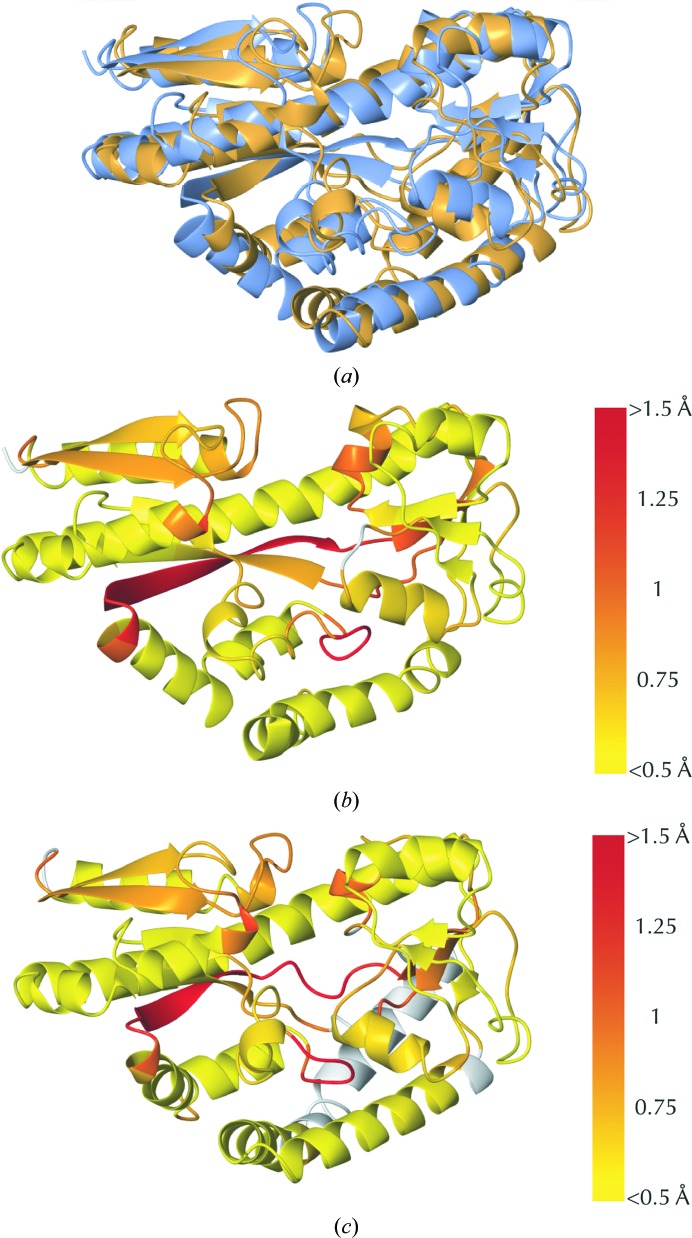
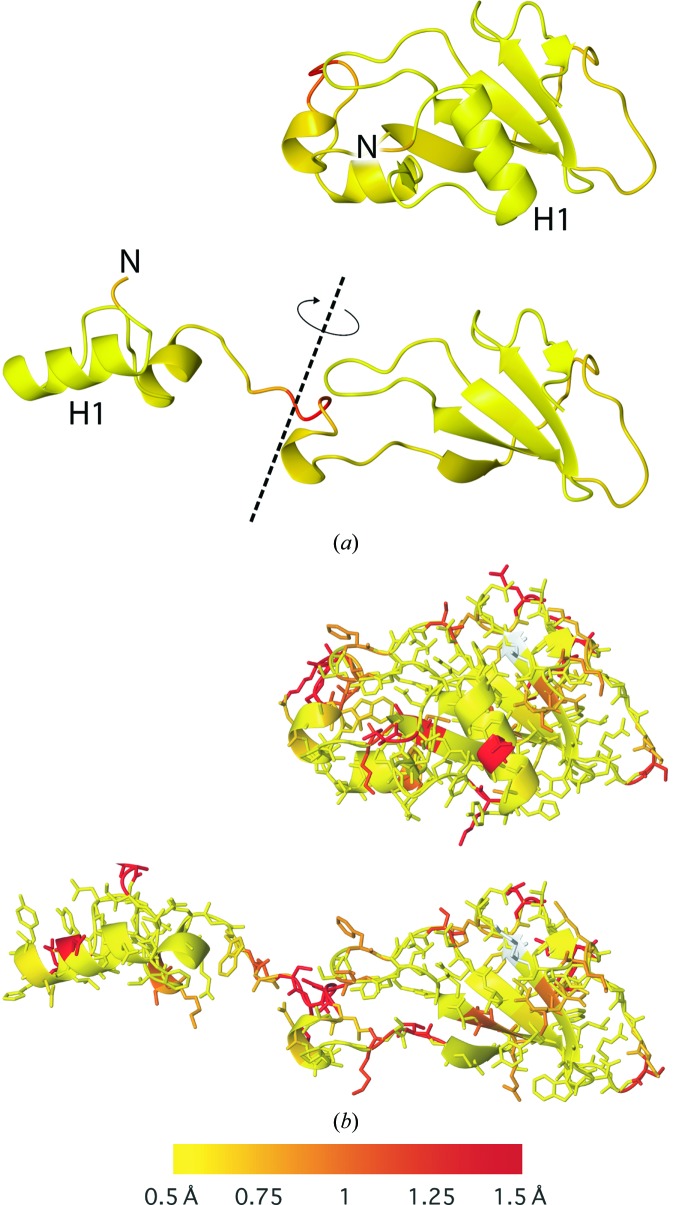
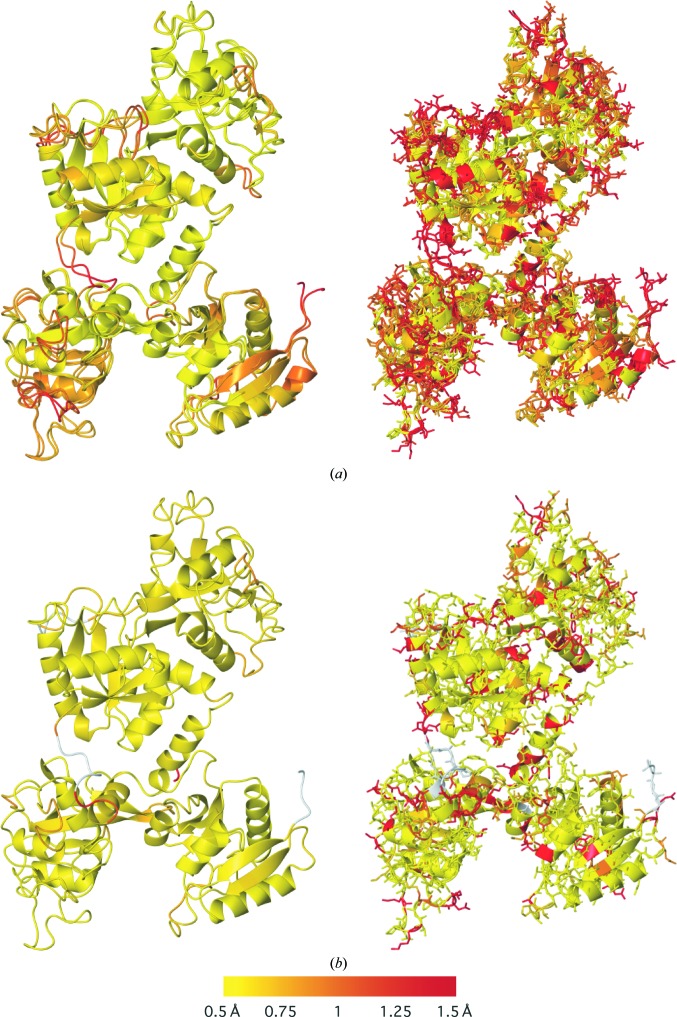
Similar articles
-
Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions.Acta Crystallogr D Biol Crystallogr. 2015 Jan 1;71(Pt 1):136-53. doi: 10.1107/S1399004714021683. Epub 2015 Jan 1. Acta Crystallogr D Biol Crystallogr. 2015. PMID: 25615868 Free PMC article.
-
Overview of refinement procedures within REFMAC5: utilizing data from different sources.Acta Crystallogr D Struct Biol. 2018 Mar 1;74(Pt 3):215-227. doi: 10.1107/S2059798318000979. Epub 2018 Mar 2. Acta Crystallogr D Struct Biol. 2018. PMID: 29533229 Free PMC article.
-
Macromolecular crowding: chemistry and physics meet biology (Ascona, Switzerland, 10-14 June 2012).Phys Biol. 2013 Aug;10(4):040301. doi: 10.1088/1478-3975/10/4/040301. Epub 2013 Aug 2. Phys Biol. 2013. PMID: 23912807
-
Low Resolution Refinement of Atomic Models Against Crystallographic Data.Methods Mol Biol. 2017;1607:565-593. doi: 10.1007/978-1-4939-7000-1_23. Methods Mol Biol. 2017. PMID: 28573589 Review.
-
Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM.Acta Crystallogr D Struct Biol. 2018 Jun 1;74(Pt 6):492-505. doi: 10.1107/S2059798318007313. Epub 2018 May 30. Acta Crystallogr D Struct Biol. 2018. PMID: 29872001 Free PMC article. Review.
Cited by
-
VASH1-SVBP and VASH2-SVBP generate different detyrosination profiles on microtubules.J Cell Biol. 2023 Feb 6;222(2):e202205096. doi: 10.1083/jcb.202205096. Epub 2022 Dec 13. J Cell Biol. 2023. PMID: 36512346 Free PMC article.
-
Structural insight into cap-snatching and RNA synthesis by influenza polymerase.Nature. 2014 Dec 18;516(7531):361-6. doi: 10.1038/nature14009. Epub 2014 Nov 19. Nature. 2014. PMID: 25409151
-
Towards Consistency in Geometry Restraints for Carbohydrates in the Pyranose form: Modern Dictionary Generators Reviewed.Curr Med Chem. 2022;29(7):1193-1207. doi: 10.2174/0929867328666210902140754. Curr Med Chem. 2022. PMID: 34477506 Free PMC article.
-
Structural basis for ligand capture and release by the endocytic receptor ApoER2.EMBO Rep. 2017 Jun;18(6):982-999. doi: 10.15252/embr.201643521. Epub 2017 Apr 26. EMBO Rep. 2017. PMID: 28446613 Free PMC article.
-
Structure of phycobilisome from the red alga Griffithsia pacifica.Nature. 2017 Nov 2;551(7678):57-63. doi: 10.1038/nature24278. Epub 2017 Oct 18. Nature. 2017. PMID: 29045394
References
-
- Alexandrov, N. N., Takahashi, K. & Go, N. (1992). J. Mol. Biol. 225, 5–9. - PubMed
-
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. (1990). J. Mol. Biol. 215, 403–410. - PubMed
-
- Aung, Z. & Tan, K. L. (2006). J. Bioinform. Comput. Biol. 4, 1197–1216. - PubMed
-
- Berman, H. et al. (2002). Acta Cryst. D58, 899–907. - PubMed
-
- Bertsekas, D. (2005). Dynamic Programming and Optimal Control, Vol 1. Nashua: Athena Scientific.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
