

β-catenin contributes to lung tumor development induced by EGFR mutations
- PMID: 25164010
- PMCID: PMC4199914
- DOI: 10.1158/0008-5472.CAN-14-0184
β-catenin contributes to lung tumor development induced by EGFR mutations
Abstract
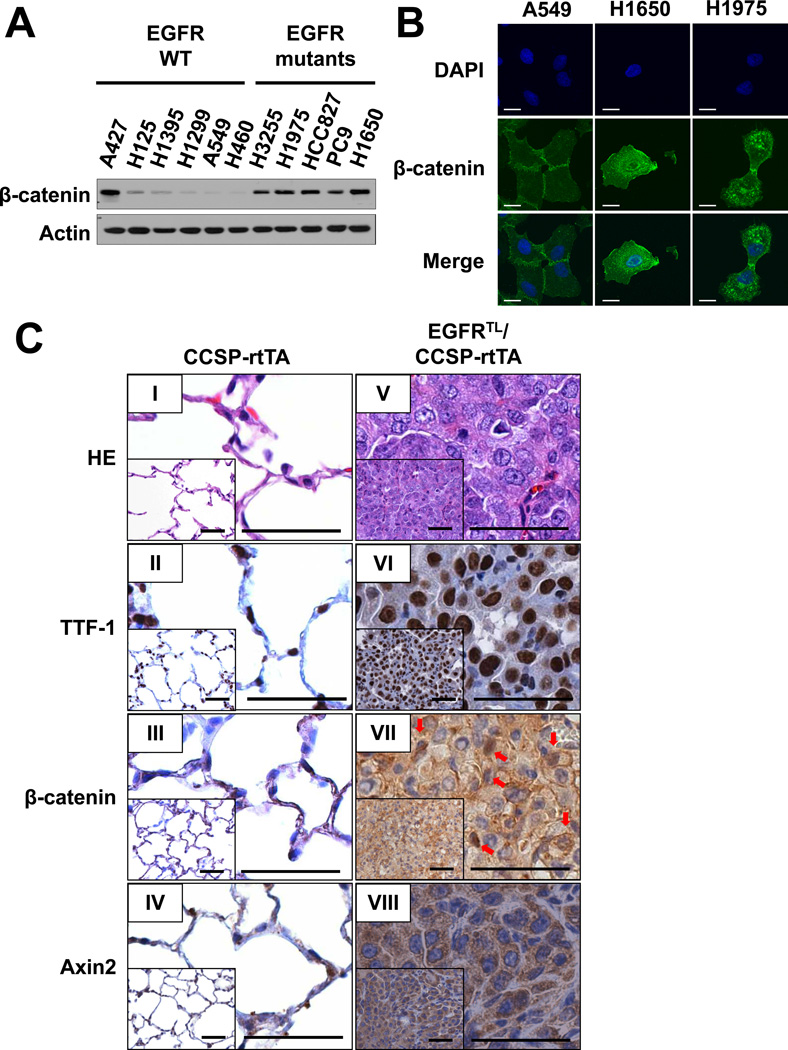
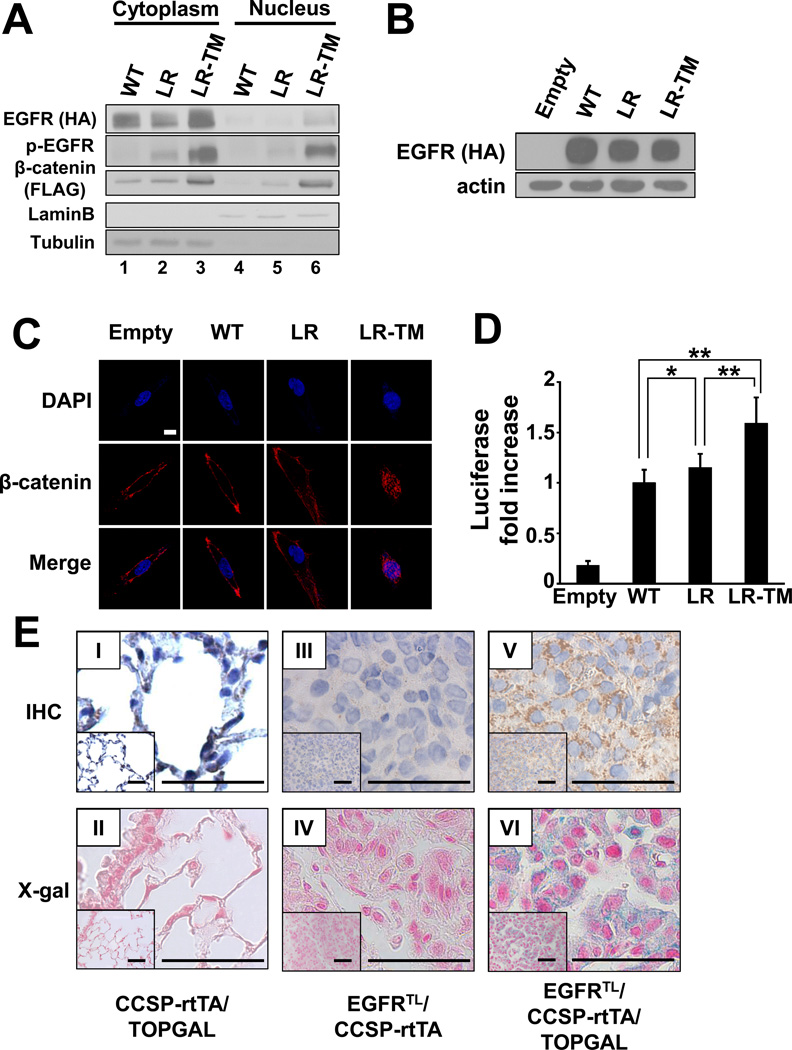
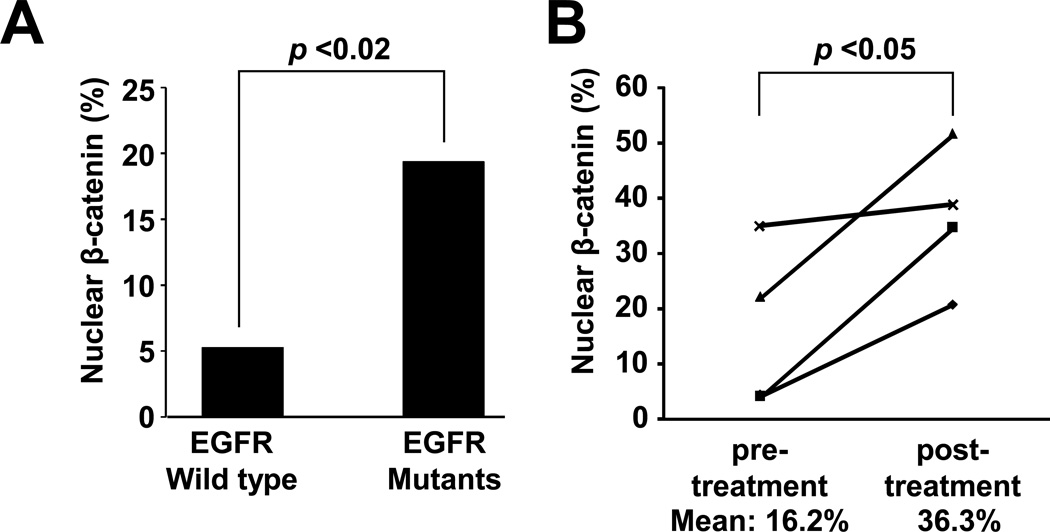
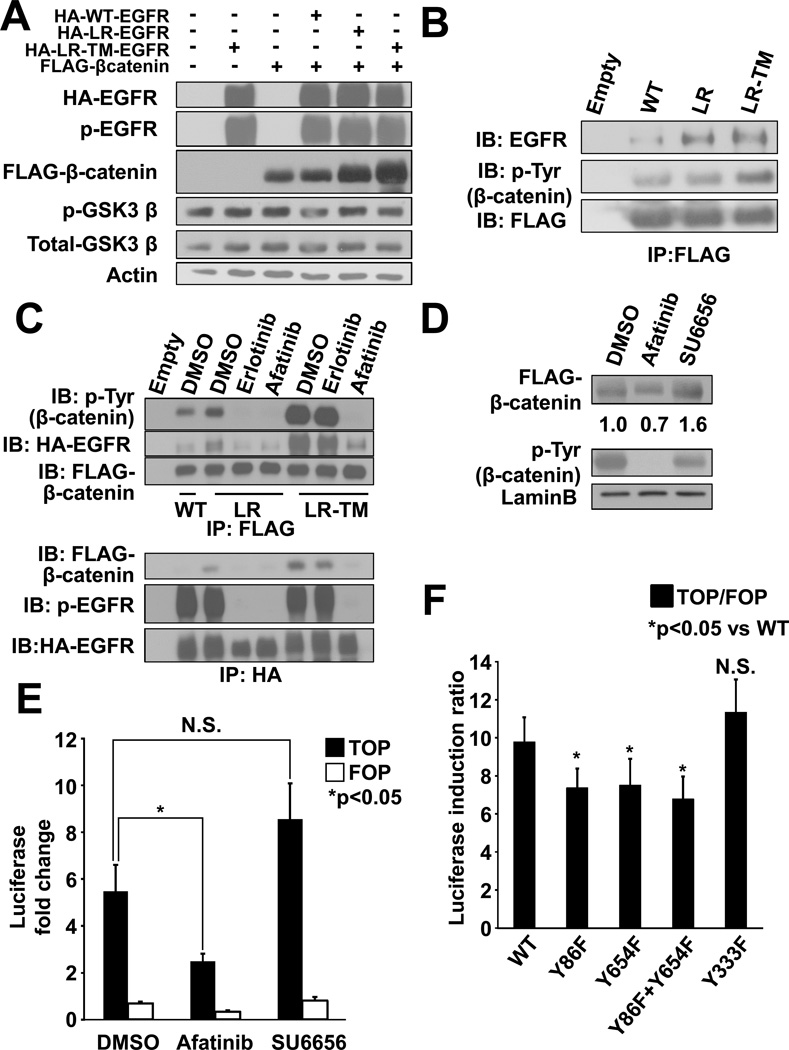
The discovery of somatic mutations in EGFR and development of EGFR tyrosine kinase inhibitors (TKI) have revolutionized treatment for lung cancer. However, resistance to TKIs emerges in almost all patients and currently no effective treatment is available. Here, we show that β-catenin is essential for development of EGFR-mutated lung cancers. β-Catenin was upregulated and activated in EGFR-mutated cells. Mutant EGFR preferentially bound to and tyrosine phosphorylated β-catenin, leading to an increase in β-catenin-mediated transactivation, particularly in cells harboring the gefitinib/erlotinib-resistant gatekeeper EGFR-T790M mutation. Pharmacologic inhibition of β-catenin suppressed EGFR-L858R-T790M mutated lung tumor growth, and genetic deletion of the β-catenin gene dramatically reduced lung tumor formation in EGFR-L858R-T790M transgenic mice. These data suggest that β-catenin plays an essential role in lung tumorigenesis and that targeting the β-catenin pathway may provide novel strategies to prevent lung cancer development or overcome resistance to EGFR TKIs.
©2014 American Association for Cancer Research.
Conflict of interest statement
D.L. is currently an employee of Pfizer. D.B.C has previously received consulting fees from AstraZeneca, Roche and Pfizer.
Figures
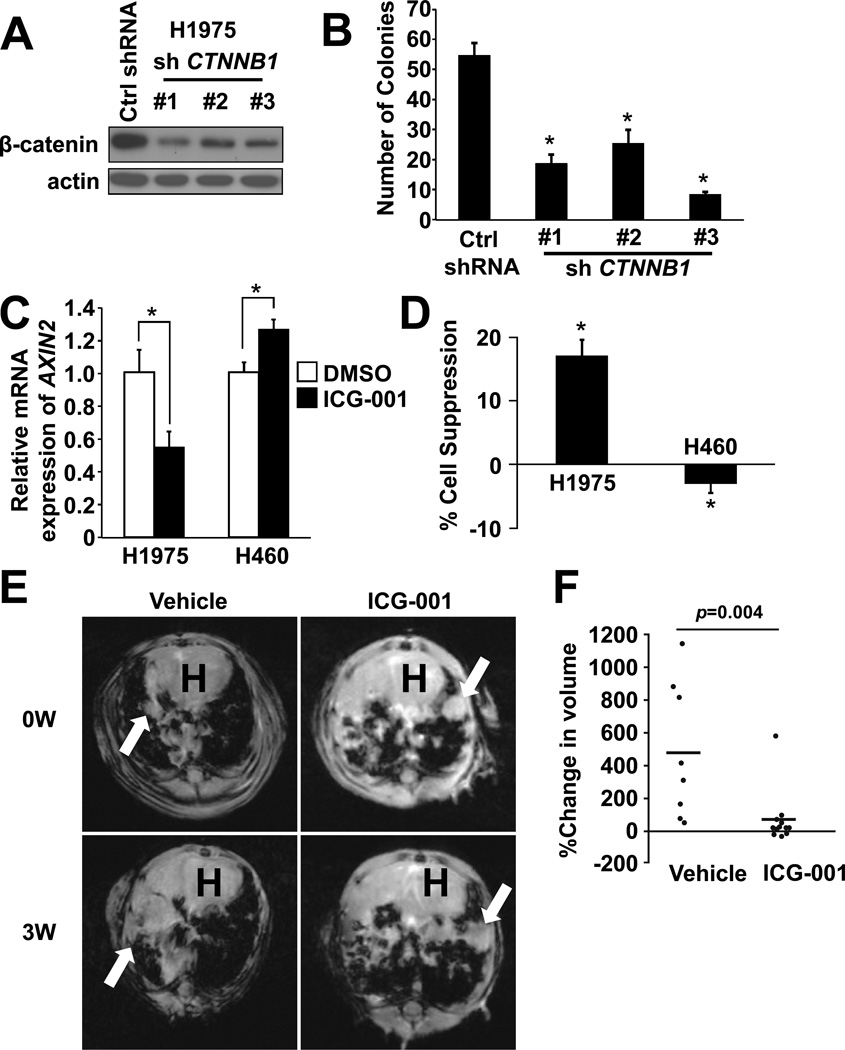
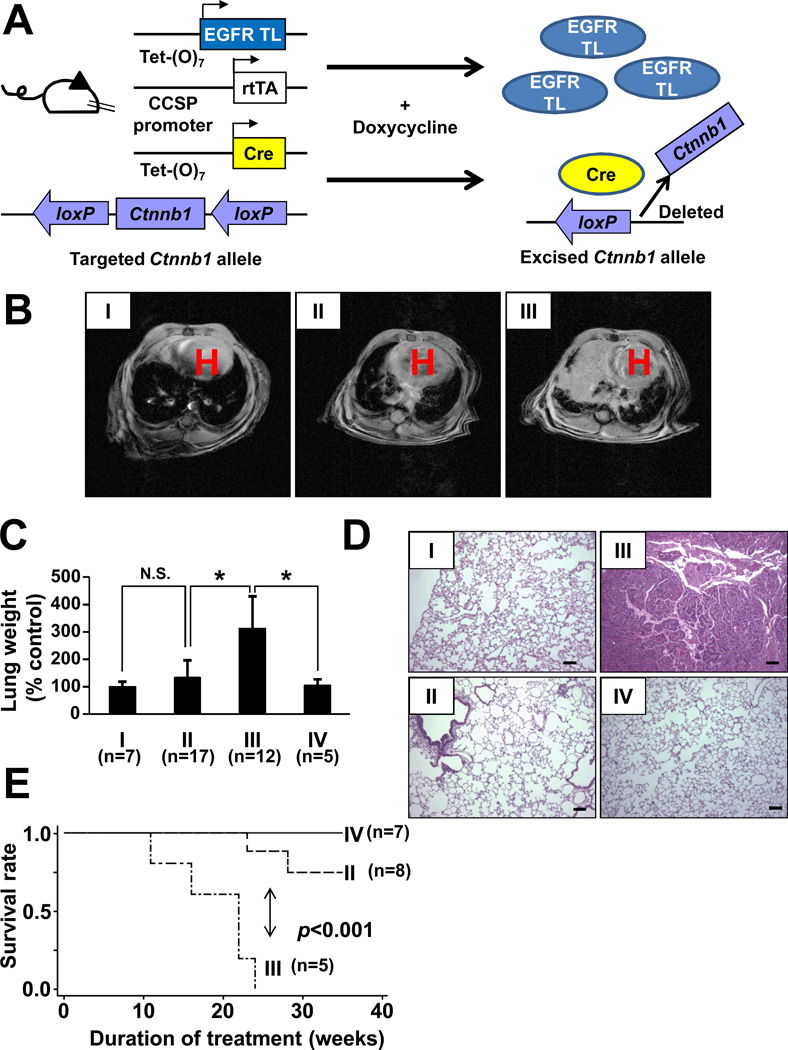
Similar articles
-
Inhibition of IGF1R signaling abrogates resistance to afatinib (BIBW2992) in EGFR T790M mutant lung cancer cells.Mol Carcinog. 2016 May;55(5):991-1001. doi: 10.1002/mc.22342. Epub 2015 Jun 4. Mol Carcinog. 2016. PMID: 26052929 Free PMC article.
-
BIM mediates EGFR tyrosine kinase inhibitor-induced apoptosis in lung cancers with oncogenic EGFR mutations.PLoS Med. 2007 Oct;4(10):1669-79; discussion 1680. doi: 10.1371/journal.pmed.0040315. PLoS Med. 2007. PMID: 17973572 Free PMC article.
-
Lower gefitinib dose led to earlier resistance acquisition before emergence of T790M mutation in epidermal growth factor receptor-mutated lung cancer model.Cancer Sci. 2013 Nov;104(11):1440-6. doi: 10.1111/cas.12284. Epub 2013 Oct 25. Cancer Sci. 2013. PMID: 24033722 Free PMC article.
-
Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancers dependent on the epidermal growth factor receptor pathway.Clin Lung Cancer. 2009 Jul;10(4):281-9. doi: 10.3816/CLC.2009.n.039. Clin Lung Cancer. 2009. PMID: 19632948 Free PMC article. Review.
-
Management and future directions in non-small cell lung cancer with known activating mutations.Am Soc Clin Oncol Educ Book. 2014:e353-65. doi: 10.14694/EdBook_AM.2014.34.e353. Am Soc Clin Oncol Educ Book. 2014. PMID: 24857124 Review.
Cited by
-
Implementation of CRISPR/Cas9 Genome Editing to Generate Murine Lung Cancer Models That Depict the Mutational Landscape of Human Disease.Front Cell Dev Biol. 2021 Mar 2;9:641618. doi: 10.3389/fcell.2021.641618. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33738287 Free PMC article.
-
Comprehensive Characterization of Molecular Differences in Cancer between Male and Female Patients.Cancer Cell. 2016 May 9;29(5):711-722. doi: 10.1016/j.ccell.2016.04.001. Cancer Cell. 2016. PMID: 27165743 Free PMC article.
-
Impact of viral presence in tumor on gene expression in non-small cell lung cancer.BMC Cancer. 2018 Aug 22;18(1):843. doi: 10.1186/s12885-018-4748-0. BMC Cancer. 2018. PMID: 30134863 Free PMC article.
-
EGFR/c-Met and mTOR signaling are predictors of survival in non-small cell lung cancer.Ther Adv Med Oncol. 2020 Sep 14;12:1758835920953731. doi: 10.1177/1758835920953731. eCollection 2020. Ther Adv Med Oncol. 2020. PMID: 32973931 Free PMC article.
-
Impact of concurrent genomic alterations in epidermal growth factor receptor (EGFR)-mutated lung cancer.J Thorac Dis. 2020 May;12(5):2883-2895. doi: 10.21037/jtd.2020.03.78. J Thorac Dis. 2020. PMID: 32642201 Free PMC article. Review.
References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. Vol. 63. CA: a cancer journal for clinicians; 2013. pp. 11–30. - PubMed
-
- Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong KK, Tenen DG, et al. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer research. 2005;65:7096–7101. - PubMed
-
- Sequist LV, Besse B, Lynch TJ, Miller VA, Wong KK, Gitlitz B, et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: results of a phase II trial in patients with advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28:3076–3083. - PubMed
-
- Miller VA, Hirsh V, Cadranel J, Chen YM, Park K, Kim SW, et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. The lancet oncology. 2012;13:528–538. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R00 CA126026/CA/NCI NIH HHS/United States
- P50CA090578/CA/NCI NIH HHS/United States
- CA166480/CA/NCI NIH HHS/United States
- R01 CA122794/CA/NCI NIH HHS/United States
- CA154303/CA/NCI NIH HHS/United States
- R01 CA169259/CA/NCI NIH HHS/United States
- R01CA169259/CA/NCI NIH HHS/United States
- P01 CA120964/CA/NCI NIH HHS/United States
- R01CA122794/CA/NCI NIH HHS/United States
- CA163896/CA/NCI NIH HHS/United States
- P01 CA154303/CA/NCI NIH HHS/United States
- R01 CA163896/CA/NCI NIH HHS/United States
- R01 CA166480/CA/NCI NIH HHS/United States
- R00CA126026/CA/NCI NIH HHS/United States
- R01 CA140594/CA/NCI NIH HHS/United States
- CA140594/CA/NCI NIH HHS/United States
- CA120964/CA/NCI NIH HHS/United States
- P50 CA090578/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
